This function performs a standard single-cell analysis workflow.
Usage
Standard_SCP(
srt,
prefix = "Standard",
assay = NULL,
do_normalization = NULL,
normalization_method = "LogNormalize",
do_HVF_finding = TRUE,
HVF_method = "vst",
nHVF = 2000,
HVF = NULL,
do_scaling = TRUE,
vars_to_regress = NULL,
regression_model = "linear",
linear_reduction = "pca",
linear_reduction_dims = 50,
linear_reduction_dims_use = NULL,
linear_reduction_params = list(),
force_linear_reduction = FALSE,
nonlinear_reduction = "umap",
nonlinear_reduction_dims = c(2, 3),
nonlinear_reduction_params = list(),
force_nonlinear_reduction = TRUE,
neighbor_metric = "euclidean",
neighbor_k = 20L,
cluster_algorithm = "louvain",
cluster_resolution = 0.6,
seed = 11
)Arguments
- srt
A Seurat object.
- prefix
A prefix to add to the names of intermediate objects created by the function (default is "Standard").
- assay
The name of the assay to use for the analysis. If NULL, the default assay of the Seurat object will be used.
- do_normalization
A logical value indicating whether to perform normalization. If NULL, normalization will be performed if the specified assay does not have scaled data.
- normalization_method
The method to use for normalization. Options are "LogNormalize", "SCT", or "TFIDF" (default is "LogNormalize").
- do_HVF_finding
A logical value indicating whether to perform high variable feature finding. If TRUE, the function will force to find the highly variable features (HVF) using the specified HVF method.
- HVF_method
The method to use for finding highly variable features. Options are "vst", "mvp" or "disp" (default is "vst").
- nHVF
The number of highly variable features to select. If NULL, all highly variable features will be used.
- HVF
A vector of feature names to use as highly variable features. If NULL, the function will use the highly variable features identified by the HVF method.
- do_scaling
A logical value indicating whether to perform scaling. If TRUE, the function will force to scale the data using the ScaleData function.
- vars_to_regress
A vector of feature names to use as regressors in the scaling step. If NULL, no regressors will be used.
- regression_model
The regression model to use for scaling. Options are "linear", "poisson", or "negativebinomial" (default is "linear").
- linear_reduction
The linear dimensionality reduction method to use. Options are "pca", "svd", "ica", "nmf", "mds", or "glmpca" (default is "pca").
- linear_reduction_dims
The number of dimensions to keep after linear dimensionality reduction (default is 50).
- linear_reduction_dims_use
The dimensions to use for downstream analysis. If NULL, all dimensions will be used.
- linear_reduction_params
A list of parameters to pass to the linear dimensionality reduction method.
- force_linear_reduction
A logical value indicating whether to force linear dimensionality reduction even if the specified reduction is already present in the Seurat object.
- nonlinear_reduction
The nonlinear dimensionality reduction method to use. Options are "umap","umap-naive", "tsne", "dm", "phate", "pacmap", "trimap", "largevis", or "fr" (default is "umap").
- nonlinear_reduction_dims
The number of dimensions to keep after nonlinear dimensionality reduction. If a vector is provided, different numbers of dimensions can be specified for each method (default is c(2, 3)).
- nonlinear_reduction_params
A list of parameters to pass to the nonlinear dimensionality reduction method.
- force_nonlinear_reduction
A logical value indicating whether to force nonlinear dimensionality reduction even if the specified reduction is already present in the Seurat object.
- neighbor_metric
The distance metric to use for finding neighbors. Options are "euclidean", "cosine", "manhattan", or "hamming" (default is "euclidean").
- neighbor_k
The number of nearest neighbors to use for finding neighbors (default is 20).
- cluster_algorithm
The clustering algorithm to use. Options are "louvain", "slm", or "leiden" (default is "louvain").
- cluster_resolution
The resolution parameter to use for clustering. Larger values result in fewer clusters (default is 0.6).
- seed
The random seed to use for reproducibility (default is 11).
Examples
data("pancreas_sub")
pancreas_sub <- Standard_SCP(pancreas_sub)
#> [2023-11-21 07:52:44.147793] Start Standard_SCP
#> [2023-11-21 07:52:44.147981] Checking srtList... ...
#> Data 1/1 of the srtList is raw_counts. Perform NormalizeData(LogNormalize) on the data ...
#> Perform FindVariableFeatures on the data 1/1 of the srtList...
#> Use the separate HVF from srtList...
#> Number of available HVF: 2000
#> [2023-11-21 07:52:44.756936] Finished checking.
#> [2023-11-21 07:52:44.757107] Perform ScaleData on the data...
#> [2023-11-21 07:52:44.831319] Perform linear dimension reduction (pca) on the data...
#> Warning: The following arguments are not used: force.recalc
#> Warning: The following arguments are not used: force.recalc
#> [2023-11-21 07:52:45.401104] Perform FindClusters (louvain) on the data...
#> [2023-11-21 07:52:45.476794] Reorder clusters...
#> [2023-11-21 07:52:45.538695] Perform nonlinear dimension reduction (umap) on the data...
#> Non-linear dimensionality reduction(umap) using Reduction(Standardpca, dims:1-13) as input
#> Found more than one class "dist" in cache; using the first, from namespace 'BiocGenerics'
#> Also defined by ‘spam’
#> Found more than one class "dist" in cache; using the first, from namespace 'BiocGenerics'
#> Also defined by ‘spam’
#> Non-linear dimensionality reduction(umap) using Reduction(Standardpca, dims:1-13) as input
#> Found more than one class "dist" in cache; using the first, from namespace 'BiocGenerics'
#> Also defined by ‘spam’
#> Found more than one class "dist" in cache; using the first, from namespace 'BiocGenerics'
#> Also defined by ‘spam’
#> [2023-11-21 07:52:53.75532] Standard_SCP done
#> Elapsed time: 9.61 secs
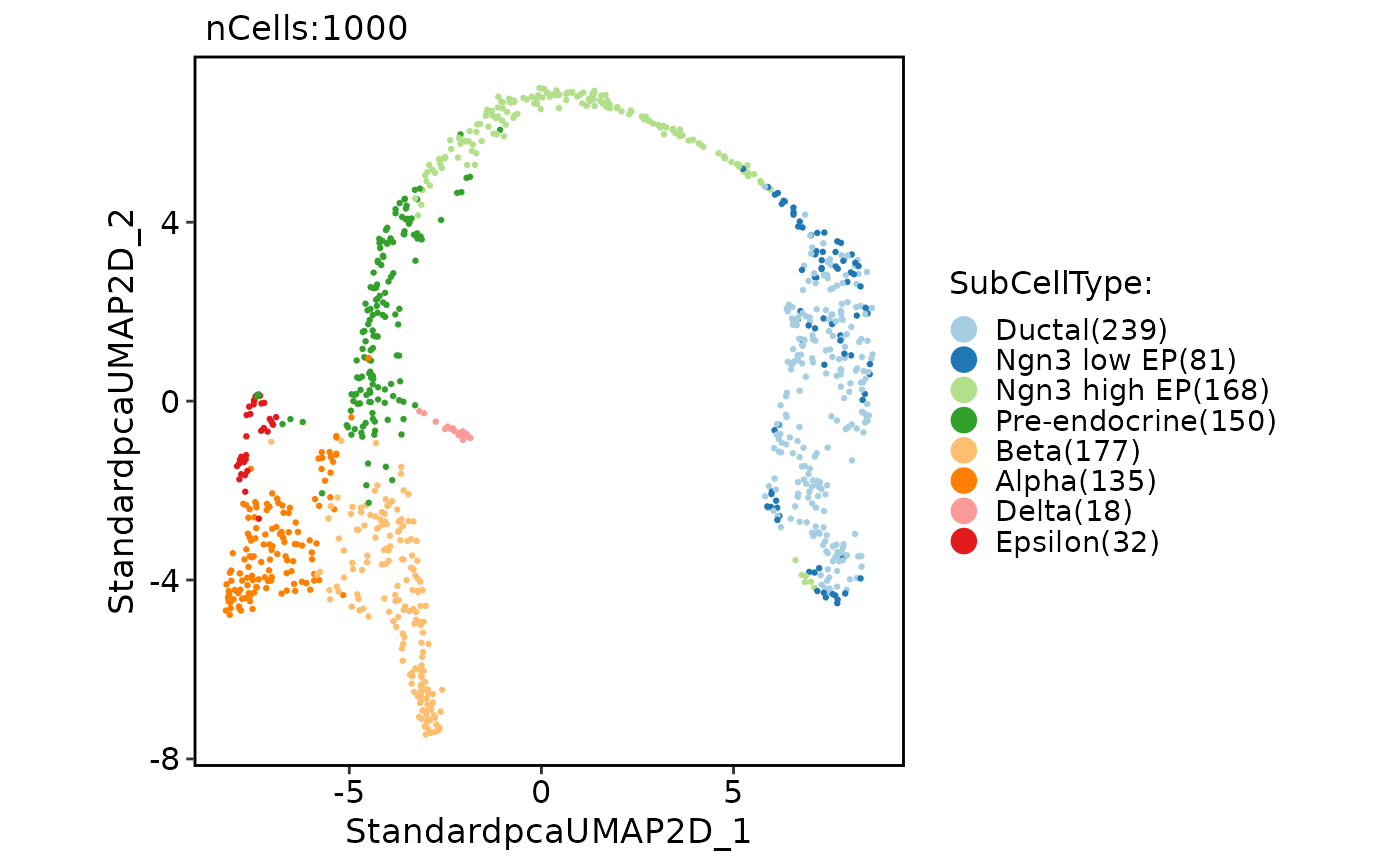
CellDimPlot(pancreas_sub, group.by = "SubCellType")
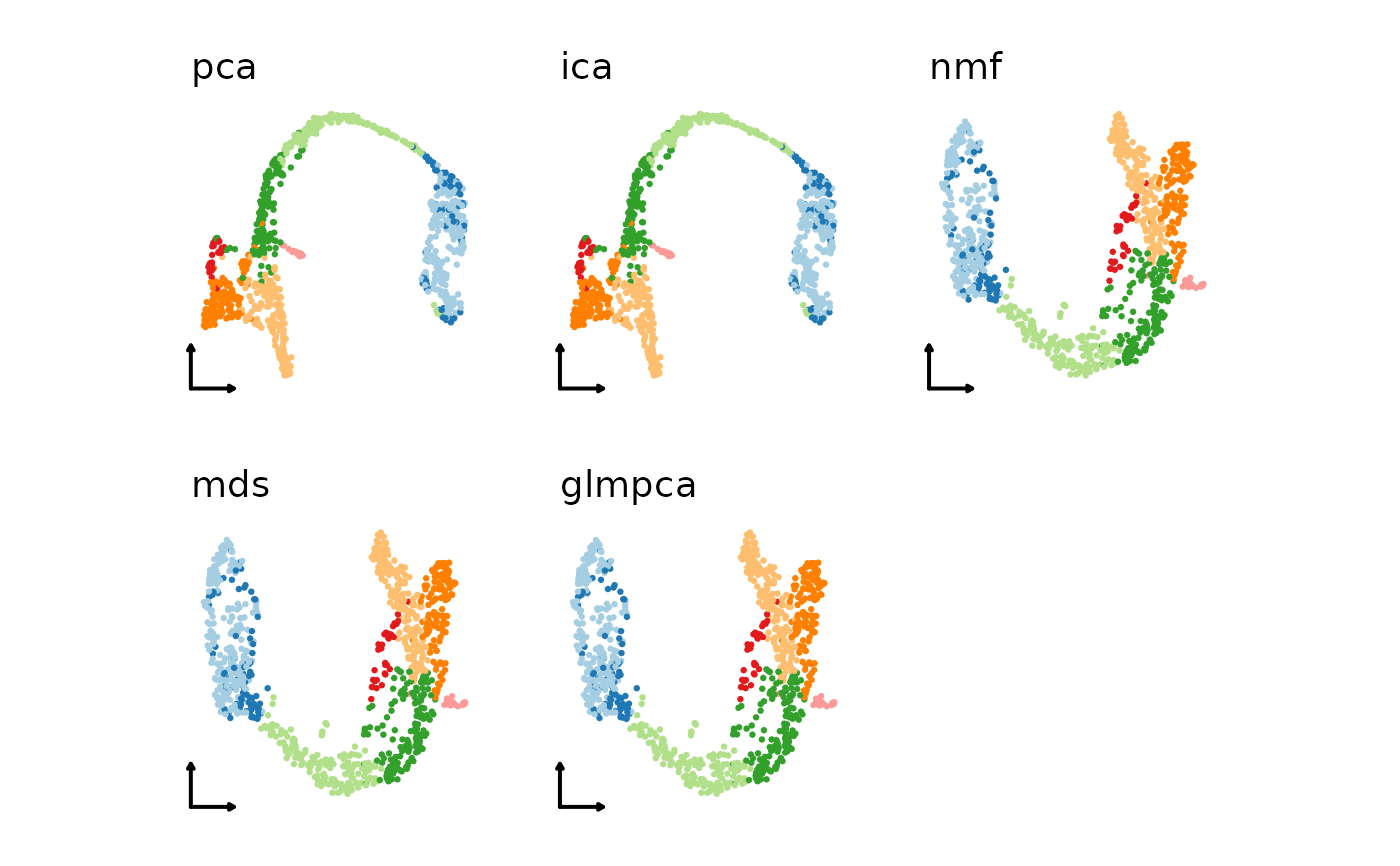
 # Use a combination of different linear or non-linear dimension reduction methods
linear_reductions <- c("pca", "ica", "nmf", "mds", "glmpca")
pancreas_sub <- Standard_SCP(
pancreas_sub,
linear_reduction = linear_reductions,
nonlinear_reduction = "umap"
)
#> [2023-11-21 07:52:53.939783] Start Standard_SCP
#> [2023-11-21 07:52:53.939937] Checking srtList... ...
#> Data 1/1 of the srtList has been log-normalized.
#> Perform FindVariableFeatures on the data 1/1 of the srtList...
#> Use the separate HVF from srtList...
#> Number of available HVF: 2000
#> [2023-11-21 07:52:54.468408] Finished checking.
#> [2023-11-21 07:52:54.468563] Perform ScaleData on the data...
#> [2023-11-21 07:52:54.552205] Perform linear dimension reduction (pca) on the data...
#> Warning: The following arguments are not used: force.recalc
#> Warning: The following arguments are not used: force.recalc
#> [2023-11-21 07:52:55.135094] Perform FindClusters (louvain) on the data...
#> [2023-11-21 07:52:55.211974] Reorder clusters...
#> [2023-11-21 07:52:55.275863] Perform nonlinear dimension reduction (umap) on the data...
#> Non-linear dimensionality reduction(umap) using Reduction(Standardpca, dims:1-13) as input
#> Found more than one class "dist" in cache; using the first, from namespace 'BiocGenerics'
#> Also defined by ‘spam’
#> Found more than one class "dist" in cache; using the first, from namespace 'BiocGenerics'
#> Also defined by ‘spam’
#> Warning: Key ‘StandardpcaUMAP2D_’ taken, using ‘standardpcaumap2d_’ instead
#> Non-linear dimensionality reduction(umap) using Reduction(Standardpca, dims:1-13) as input
#> Found more than one class "dist" in cache; using the first, from namespace 'BiocGenerics'
#> Also defined by ‘spam’
#> Found more than one class "dist" in cache; using the first, from namespace 'BiocGenerics'
#> Also defined by ‘spam’
#> Warning: Key ‘StandardpcaUMAP3D_’ taken, using ‘standardpcaumap3d_’ instead
#> [2023-11-21 07:53:03.459042] Perform linear dimension reduction (ica) on the data...
#> Error in validObject(.Object): invalid class “DimReduc” object: rownames must be present in 'cell.embeddings'
plist1 <- lapply(linear_reductions, function(lr) {
CellDimPlot(pancreas_sub,
group.by = "SubCellType",
reduction = paste0("Standard", lr, "UMAP2D"),
xlab = "", ylab = "", title = lr,
legend.position = "none",
theme_use = "theme_blank"
)
})
patchwork::wrap_plots(plotlist = plist1)
# Use a combination of different linear or non-linear dimension reduction methods
linear_reductions <- c("pca", "ica", "nmf", "mds", "glmpca")
pancreas_sub <- Standard_SCP(
pancreas_sub,
linear_reduction = linear_reductions,
nonlinear_reduction = "umap"
)
#> [2023-11-21 07:52:53.939783] Start Standard_SCP
#> [2023-11-21 07:52:53.939937] Checking srtList... ...
#> Data 1/1 of the srtList has been log-normalized.
#> Perform FindVariableFeatures on the data 1/1 of the srtList...
#> Use the separate HVF from srtList...
#> Number of available HVF: 2000
#> [2023-11-21 07:52:54.468408] Finished checking.
#> [2023-11-21 07:52:54.468563] Perform ScaleData on the data...
#> [2023-11-21 07:52:54.552205] Perform linear dimension reduction (pca) on the data...
#> Warning: The following arguments are not used: force.recalc
#> Warning: The following arguments are not used: force.recalc
#> [2023-11-21 07:52:55.135094] Perform FindClusters (louvain) on the data...
#> [2023-11-21 07:52:55.211974] Reorder clusters...
#> [2023-11-21 07:52:55.275863] Perform nonlinear dimension reduction (umap) on the data...
#> Non-linear dimensionality reduction(umap) using Reduction(Standardpca, dims:1-13) as input
#> Found more than one class "dist" in cache; using the first, from namespace 'BiocGenerics'
#> Also defined by ‘spam’
#> Found more than one class "dist" in cache; using the first, from namespace 'BiocGenerics'
#> Also defined by ‘spam’
#> Warning: Key ‘StandardpcaUMAP2D_’ taken, using ‘standardpcaumap2d_’ instead
#> Non-linear dimensionality reduction(umap) using Reduction(Standardpca, dims:1-13) as input
#> Found more than one class "dist" in cache; using the first, from namespace 'BiocGenerics'
#> Also defined by ‘spam’
#> Found more than one class "dist" in cache; using the first, from namespace 'BiocGenerics'
#> Also defined by ‘spam’
#> Warning: Key ‘StandardpcaUMAP3D_’ taken, using ‘standardpcaumap3d_’ instead
#> [2023-11-21 07:53:03.459042] Perform linear dimension reduction (ica) on the data...
#> Error in validObject(.Object): invalid class “DimReduc” object: rownames must be present in 'cell.embeddings'
plist1 <- lapply(linear_reductions, function(lr) {
CellDimPlot(pancreas_sub,
group.by = "SubCellType",
reduction = paste0("Standard", lr, "UMAP2D"),
xlab = "", ylab = "", title = lr,
legend.position = "none",
theme_use = "theme_blank"
)
})
patchwork::wrap_plots(plotlist = plist1)
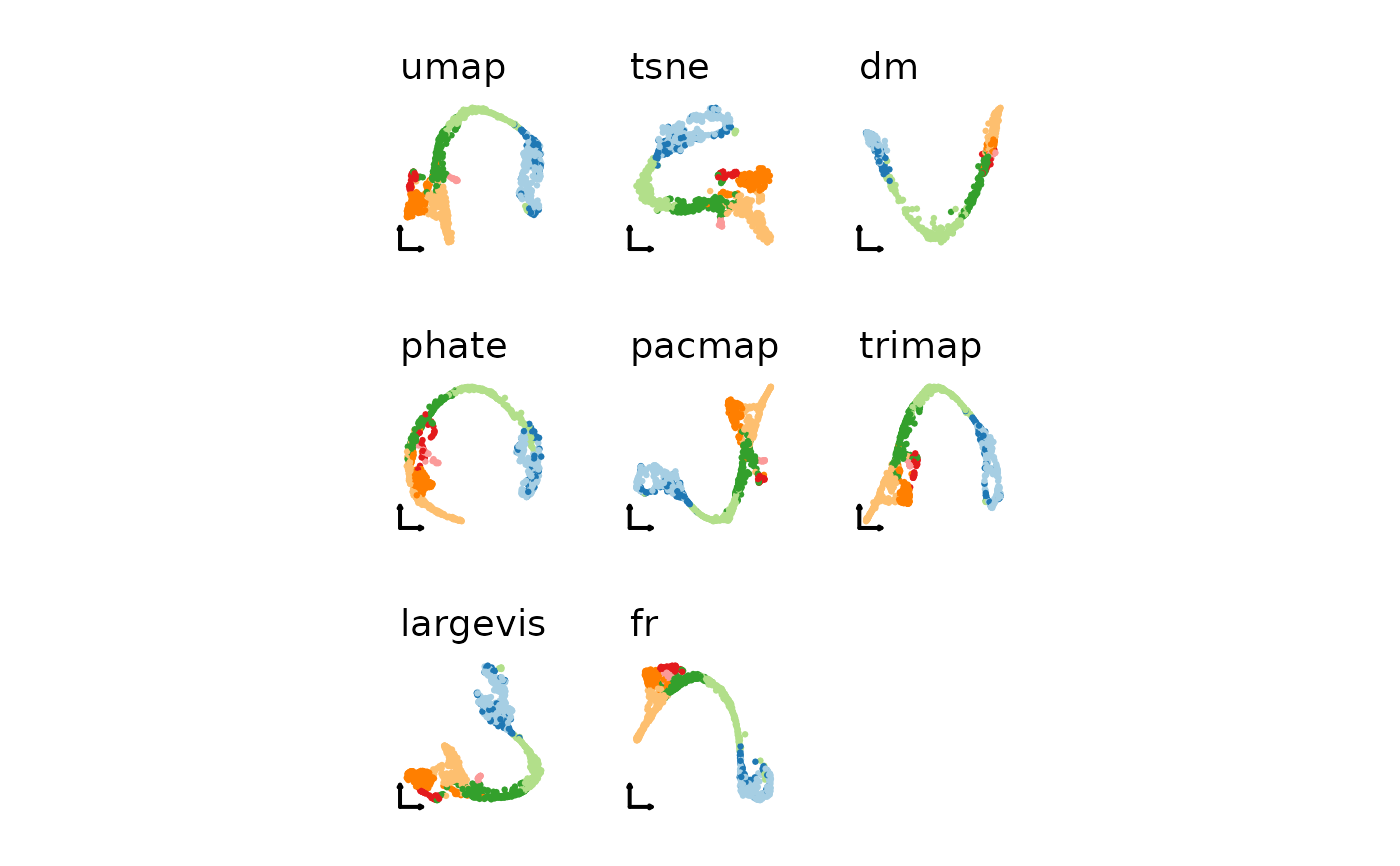
 nonlinear_reductions <- c("umap", "tsne", "dm", "phate", "pacmap", "trimap", "largevis", "fr")
pancreas_sub <- Standard_SCP(
pancreas_sub,
linear_reduction = "pca",
nonlinear_reduction = nonlinear_reductions
)
#> [2023-11-21 07:53:04.813962] Start Standard_SCP
#> [2023-11-21 07:53:04.814099] Checking srtList... ...
#> Data 1/1 of the srtList has been log-normalized.
#> Perform FindVariableFeatures on the data 1/1 of the srtList...
#> Use the separate HVF from srtList...
#> Number of available HVF: 2000
#> [2023-11-21 07:53:05.368413] Finished checking.
#> [2023-11-21 07:53:05.368597] Perform ScaleData on the data...
#> [2023-11-21 07:53:05.454685] Perform linear dimension reduction (pca) on the data...
#> Warning: The following arguments are not used: force.recalc
#> Warning: The following arguments are not used: force.recalc
#> [2023-11-21 07:53:06.079364] Perform FindClusters (louvain) on the data...
#> [2023-11-21 07:53:06.157565] Reorder clusters...
#> [2023-11-21 07:53:06.222641] Perform nonlinear dimension reduction (umap) on the data...
#> Non-linear dimensionality reduction(umap) using Reduction(Standardpca, dims:1-13) as input
#> Found more than one class "dist" in cache; using the first, from namespace 'BiocGenerics'
#> Also defined by ‘spam’
#> Found more than one class "dist" in cache; using the first, from namespace 'BiocGenerics'
#> Also defined by ‘spam’
#> Warning: Key ‘StandardpcaUMAP2D_’ taken, using ‘standardpcaumap2d_’ instead
#> Non-linear dimensionality reduction(umap) using Reduction(Standardpca, dims:1-13) as input
#> Found more than one class "dist" in cache; using the first, from namespace 'BiocGenerics'
#> Also defined by ‘spam’
#> Found more than one class "dist" in cache; using the first, from namespace 'BiocGenerics'
#> Also defined by ‘spam’
#> Warning: Key ‘StandardpcaUMAP3D_’ taken, using ‘standardpcaumap3d_’ instead
#> [2023-11-21 07:53:14.479583] Perform nonlinear dimension reduction (tsne) on the data...
#> Non-linear dimensionality reduction(tsne) using Reduction(Standardpca, dims:1-13) as input
#> Non-linear dimensionality reduction(tsne) using Reduction(Standardpca, dims:1-13) as input
#> [2023-11-21 07:53:20.038773] Perform nonlinear dimension reduction (dm) on the data...
#> Non-linear dimensionality reduction(dm) using Reduction(Standardpca, dims:1-13) as input
#> Warning: In destiny::DiffusionMap(data = as_matrix(object), n_eigs = ndcs,
#> sigma = sigma, k = k, distance = dist.method, verbose = verbose,
#> ...) :
#> extra argument ‘graph’ will be disregarded
#> Non-linear dimensionality reduction(dm) using Reduction(Standardpca, dims:1-13) as input
#> Warning: In destiny::DiffusionMap(data = as_matrix(object), n_eigs = ndcs,
#> sigma = sigma, k = k, distance = dist.method, verbose = verbose,
#> ...) :
#> extra argument ‘graph’ will be disregarded
#> [2023-11-21 07:53:20.407119] Perform nonlinear dimension reduction (phate) on the data...
#> Non-linear dimensionality reduction(phate) using Reduction(Standardpca, dims:1-13) as input
#> Non-linear dimensionality reduction(phate) using Reduction(Standardpca, dims:1-13) as input
#> [2023-11-21 07:53:53.887008] Perform nonlinear dimension reduction (pacmap) on the data...
#> Non-linear dimensionality reduction(pacmap) using Reduction(Standardpca, dims:1-13) as input
#> Non-linear dimensionality reduction(pacmap) using Reduction(Standardpca, dims:1-13) as input
#> [2023-11-21 07:54:07.125435] Perform nonlinear dimension reduction (trimap) on the data...
#> Non-linear dimensionality reduction(trimap) using Reduction(Standardpca, dims:1-13) as input
#> Non-linear dimensionality reduction(trimap) using Reduction(Standardpca, dims:1-13) as input
#> [2023-11-21 07:54:21.320742] Perform nonlinear dimension reduction (largevis) on the data...
#> Non-linear dimensionality reduction(largevis) using Reduction(Standardpca, dims:1-13) as input
#> Found more than one class "dist" in cache; using the first, from namespace 'BiocGenerics'
#> Also defined by ‘spam’
#> Found more than one class "dist" in cache; using the first, from namespace 'BiocGenerics'
#> Also defined by ‘spam’
#> Non-linear dimensionality reduction(largevis) using Reduction(Standardpca, dims:1-13) as input
#> Found more than one class "dist" in cache; using the first, from namespace 'BiocGenerics'
#> Also defined by ‘spam’
#> Found more than one class "dist" in cache; using the first, from namespace 'BiocGenerics'
#> Also defined by ‘spam’
#> [2023-11-21 07:59:07.277978] Perform nonlinear dimension reduction (fr) on the data...
#> Non-linear dimensionality reduction(fr) using Graphs(Standardpca_SNN) as input
#> Non-linear dimensionality reduction(fr) using Graphs(Standardpca_SNN) as input
#> [2023-11-21 07:59:10.110768] Standard_SCP done
#> Elapsed time: 6.09 mins
plist2 <- lapply(nonlinear_reductions, function(nr) {
CellDimPlot(pancreas_sub,
group.by = "SubCellType",
reduction = paste0("Standardpca", toupper(nr), "2D"),
xlab = "", ylab = "", title = nr,
legend.position = "none",
theme_use = "theme_blank"
)
})
patchwork::wrap_plots(plotlist = plist2)
nonlinear_reductions <- c("umap", "tsne", "dm", "phate", "pacmap", "trimap", "largevis", "fr")
pancreas_sub <- Standard_SCP(
pancreas_sub,
linear_reduction = "pca",
nonlinear_reduction = nonlinear_reductions
)
#> [2023-11-21 07:53:04.813962] Start Standard_SCP
#> [2023-11-21 07:53:04.814099] Checking srtList... ...
#> Data 1/1 of the srtList has been log-normalized.
#> Perform FindVariableFeatures on the data 1/1 of the srtList...
#> Use the separate HVF from srtList...
#> Number of available HVF: 2000
#> [2023-11-21 07:53:05.368413] Finished checking.
#> [2023-11-21 07:53:05.368597] Perform ScaleData on the data...
#> [2023-11-21 07:53:05.454685] Perform linear dimension reduction (pca) on the data...
#> Warning: The following arguments are not used: force.recalc
#> Warning: The following arguments are not used: force.recalc
#> [2023-11-21 07:53:06.079364] Perform FindClusters (louvain) on the data...
#> [2023-11-21 07:53:06.157565] Reorder clusters...
#> [2023-11-21 07:53:06.222641] Perform nonlinear dimension reduction (umap) on the data...
#> Non-linear dimensionality reduction(umap) using Reduction(Standardpca, dims:1-13) as input
#> Found more than one class "dist" in cache; using the first, from namespace 'BiocGenerics'
#> Also defined by ‘spam’
#> Found more than one class "dist" in cache; using the first, from namespace 'BiocGenerics'
#> Also defined by ‘spam’
#> Warning: Key ‘StandardpcaUMAP2D_’ taken, using ‘standardpcaumap2d_’ instead
#> Non-linear dimensionality reduction(umap) using Reduction(Standardpca, dims:1-13) as input
#> Found more than one class "dist" in cache; using the first, from namespace 'BiocGenerics'
#> Also defined by ‘spam’
#> Found more than one class "dist" in cache; using the first, from namespace 'BiocGenerics'
#> Also defined by ‘spam’
#> Warning: Key ‘StandardpcaUMAP3D_’ taken, using ‘standardpcaumap3d_’ instead
#> [2023-11-21 07:53:14.479583] Perform nonlinear dimension reduction (tsne) on the data...
#> Non-linear dimensionality reduction(tsne) using Reduction(Standardpca, dims:1-13) as input
#> Non-linear dimensionality reduction(tsne) using Reduction(Standardpca, dims:1-13) as input
#> [2023-11-21 07:53:20.038773] Perform nonlinear dimension reduction (dm) on the data...
#> Non-linear dimensionality reduction(dm) using Reduction(Standardpca, dims:1-13) as input
#> Warning: In destiny::DiffusionMap(data = as_matrix(object), n_eigs = ndcs,
#> sigma = sigma, k = k, distance = dist.method, verbose = verbose,
#> ...) :
#> extra argument ‘graph’ will be disregarded
#> Non-linear dimensionality reduction(dm) using Reduction(Standardpca, dims:1-13) as input
#> Warning: In destiny::DiffusionMap(data = as_matrix(object), n_eigs = ndcs,
#> sigma = sigma, k = k, distance = dist.method, verbose = verbose,
#> ...) :
#> extra argument ‘graph’ will be disregarded
#> [2023-11-21 07:53:20.407119] Perform nonlinear dimension reduction (phate) on the data...
#> Non-linear dimensionality reduction(phate) using Reduction(Standardpca, dims:1-13) as input
#> Non-linear dimensionality reduction(phate) using Reduction(Standardpca, dims:1-13) as input
#> [2023-11-21 07:53:53.887008] Perform nonlinear dimension reduction (pacmap) on the data...
#> Non-linear dimensionality reduction(pacmap) using Reduction(Standardpca, dims:1-13) as input
#> Non-linear dimensionality reduction(pacmap) using Reduction(Standardpca, dims:1-13) as input
#> [2023-11-21 07:54:07.125435] Perform nonlinear dimension reduction (trimap) on the data...
#> Non-linear dimensionality reduction(trimap) using Reduction(Standardpca, dims:1-13) as input
#> Non-linear dimensionality reduction(trimap) using Reduction(Standardpca, dims:1-13) as input
#> [2023-11-21 07:54:21.320742] Perform nonlinear dimension reduction (largevis) on the data...
#> Non-linear dimensionality reduction(largevis) using Reduction(Standardpca, dims:1-13) as input
#> Found more than one class "dist" in cache; using the first, from namespace 'BiocGenerics'
#> Also defined by ‘spam’
#> Found more than one class "dist" in cache; using the first, from namespace 'BiocGenerics'
#> Also defined by ‘spam’
#> Non-linear dimensionality reduction(largevis) using Reduction(Standardpca, dims:1-13) as input
#> Found more than one class "dist" in cache; using the first, from namespace 'BiocGenerics'
#> Also defined by ‘spam’
#> Found more than one class "dist" in cache; using the first, from namespace 'BiocGenerics'
#> Also defined by ‘spam’
#> [2023-11-21 07:59:07.277978] Perform nonlinear dimension reduction (fr) on the data...
#> Non-linear dimensionality reduction(fr) using Graphs(Standardpca_SNN) as input
#> Non-linear dimensionality reduction(fr) using Graphs(Standardpca_SNN) as input
#> [2023-11-21 07:59:10.110768] Standard_SCP done
#> Elapsed time: 6.09 mins
plist2 <- lapply(nonlinear_reductions, function(nr) {
CellDimPlot(pancreas_sub,
group.by = "SubCellType",
reduction = paste0("Standardpca", toupper(nr), "2D"),
xlab = "", ylab = "", title = nr,
legend.position = "none",
theme_use = "theme_blank"
)
})
patchwork::wrap_plots(plotlist = plist2)