This function handles multiple quality control methods for single-cell RNA-seq data.
Usage
RunCellQC(
srt,
assay = "RNA",
split.by = NULL,
return_filtered = FALSE,
qc_metrics = c("doublets", "outlier", "umi", "gene", "mito", "ribo", "ribo_mito_ratio",
"species"),
db_method = "scDblFinder",
db_rate = NULL,
db_coefficient = 0.01,
outlier_threshold = c("log10_nCount:lower:2.5", "log10_nCount:higher:5",
"log10_nFeature:lower:2.5", "log10_nFeature:higher:5", "featurecount_dist:lower:2.5"),
outlier_n = 1,
UMI_threshold = 3000,
gene_threshold = 1000,
mito_threshold = 20,
mito_pattern = c("MT-", "Mt-", "mt-"),
mito_gene = NULL,
ribo_threshold = 50,
ribo_pattern = c("RP[SL]\\d+\\w{0,1}\\d*$", "Rp[sl]\\d+\\w{0,1}\\d*$",
"rp[sl]\\d+\\w{0,1}\\d*$"),
ribo_gene = NULL,
ribo_mito_ratio_range = c(1, Inf),
species = NULL,
species_gene_prefix = NULL,
species_percent = 95,
seed = 11
)Arguments
- srt
A Seurat object.
- assay
The name of the assay to be used for doublet-calling. Default is "RNA".
- split.by
Name of the sample variable to split the Seurat object. Default is NULL.
- return_filtered
Logical indicating whether to return a cell-filtered Seurat object. Default is FALSE.
- qc_metrics
A character vector specifying the quality control metrics to be applied. Default is `c("doublets", "outlier", "umi", "gene", "mito", "ribo", "ribo_mito_ratio", "species")`.
- db_method
Doublet-calling methods used. Can be one of
scDblFinder,Scrublet,DoubletDetection,scds_cxds,scds_bcds,scds_hybrid- db_rate
The expected doublet rate. By default this is assumed to be 1% per thousand cells captured (so 4% among 4000 thousand cells), which is appropriate for 10x datasets.
- db_coefficient
The coefficient used to calculate the doublet rate. Default is 0.01. Doublet rate is calculated as`ncol(srt) / 1000 * db_coefficient`
- outlier_threshold
A character vector specifying the outlier threshold. Default is `c("log10_nCount:lower:2.5", "log10_nCount:higher:5", "log10_nFeature:lower:2.5", "log10_nFeature:higher:5", "featurecount_dist:lower:2.5")`. See isOutlier.
- outlier_n
Minimum number of outlier metrics that meet the conditions for determining outlier cells. Default is 1.
- UMI_threshold
UMI number threshold. Cells that exceed this threshold will be considered as kept. Default is 3000.
- gene_threshold
Gene number threshold. Cells that exceed this threshold will be considered as kept. Default is 1000.
- mito_threshold
Percentage of UMI counts of mitochondrial genes. Cells that exceed this threshold will be considered as discarded. Default is 20.
- mito_pattern
Regex patterns to match the mitochondrial genes. Default is `c("MT-", "Mt-", "mt-")`.
- mito_gene
A defined mitochondrial genes. If features provided, will ignore the
mito_patternmatching. Default isNULL.- ribo_threshold
Percentage of UMI counts of ribosomal genes. Cells that exceed this threshold will be considered as discarded. Default is 50.
- ribo_pattern
Regex patterns to match the ribosomal genes. Default is `c("RP[SL]\d+\w0,1\d*$", "Rp[sl]\d+\w0,1\d*$", "rp[sl]\d+\w0,1\d*$")`.
- ribo_gene
A defined ribosomal genes. If features provided, will ignore the
ribo_patternmatching. Default isNULL.- ribo_mito_ratio_range
A numeric vector specifying the range of ribosomal/mitochondrial gene expression ratios for ribo_mito_ratio outlier cells. Default is c(1, Inf).
- species
Species used as the suffix of the QC metrics. The first is the species of interest. Default is
NULL.- species_gene_prefix
Species gene prefix used to calculate QC metrics for each species. Default is
NULL.- species_percent
Percentage of UMI counts of the first species. Cells that exceed this threshold will be considered as kept. Default is 95.
- seed
Set a random seed. Default is 11.
Examples
data("pancreas_sub")
pancreas_sub <- RunCellQC(pancreas_sub)
#> >>> Total cells: 1000
#> >>> Cells which are filtered out: 26
#> ... 12 potential doublets
#> ... 14 outlier cells
#> ... 0 low-UMI cells
#> ... 0 low-gene cells
#> ... 0 high-mito cells
#> ... 0 high-ribo cells
#> ... 0 ribo_mito_ratio outlier cells
#> ... 0 species-contaminated cells
#> >>> Remained cells after filtering: 974
CellStatPlot(
srt = pancreas_sub,
stat.by = c(
"db_qc", "outlier_qc", "umi_qc", "gene_qc",
"mito_qc", "ribo_qc", "ribo_mito_ratio_qc", "species_qc"
),
plot_type = "upset", stat_level = "Fail"
)
#> Warning: stat_type is forcibly set to 'count' when plot sankey, chord, venn or upset
#> `geom_line()`: Each group consists of only one observation.
#> ℹ Do you need to adjust the group aesthetic?
#> `geom_line()`: Each group consists of only one observation.
#> ℹ Do you need to adjust the group aesthetic?
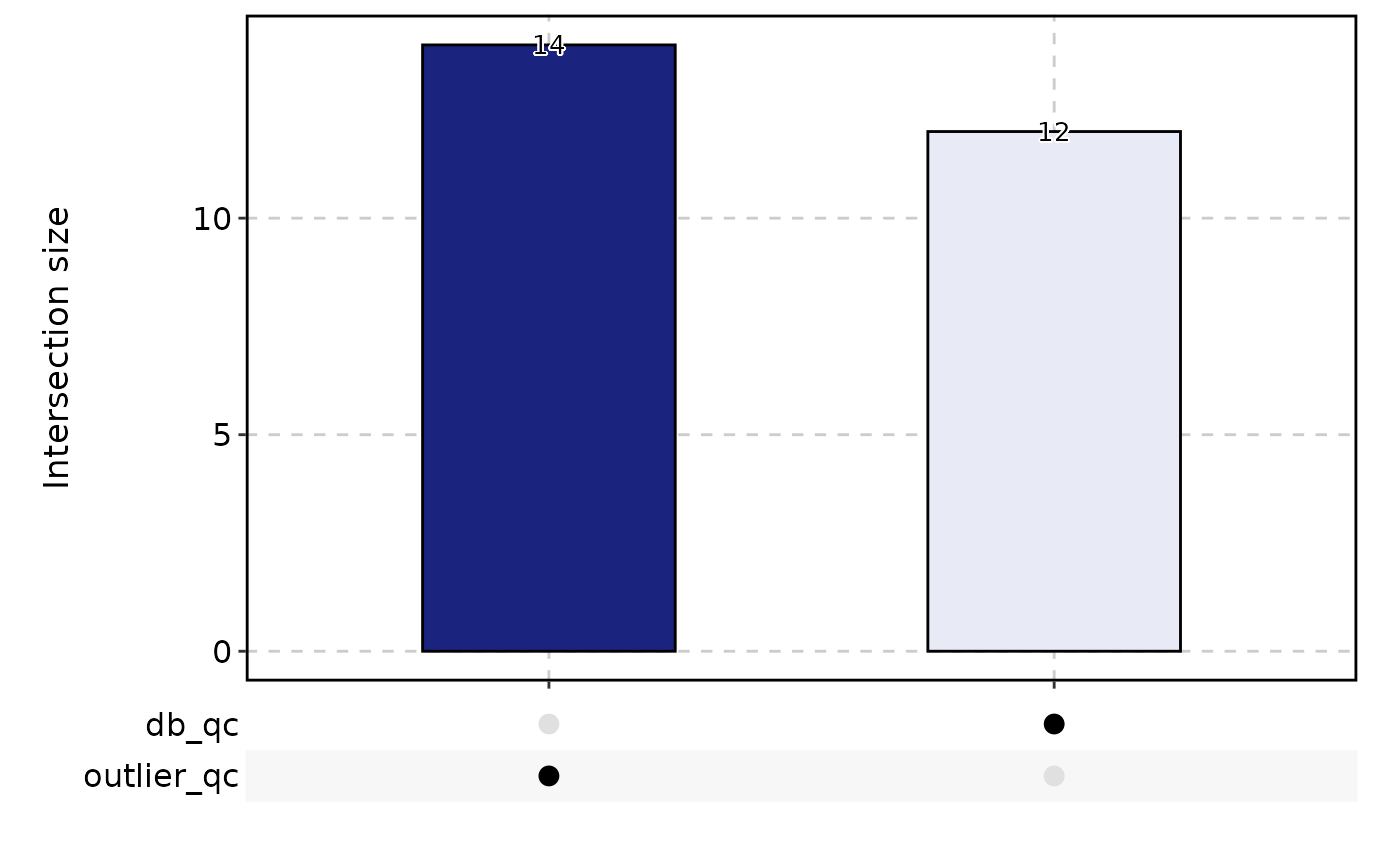 table(pancreas_sub$CellQC)
#>
#> Pass Fail
#> 974 26
data("ifnb_sub")
ifnb_sub <- RunCellQC(ifnb_sub, split.by = "stim", UMI_threshold = 1000, gene_threshold = 550)
#> === CTRL ===
#> >>> Total cells: 1000
#> >>> Cells which are filtered out: 315
#> ... 54 potential doublets
#> ... 8 outlier cells
#> ... 28 low-UMI cells
#> ... 250 low-gene cells
#> ... 0 high-mito cells
#> ... 0 high-ribo cells
#> ... 0 ribo_mito_ratio outlier cells
#> ... 0 species-contaminated cells
#> >>> Remained cells after filtering: 685
#> === STIM ===
#> >>> Total cells: 1000
#> >>> Cells which are filtered out: 303
#> ... 42 potential doublets
#> ... 12 outlier cells
#> ... 25 low-UMI cells
#> ... 251 low-gene cells
#> ... 0 high-mito cells
#> ... 0 high-ribo cells
#> ... 0 ribo_mito_ratio outlier cells
#> ... 0 species-contaminated cells
#> >>> Remained cells after filtering: 697
CellStatPlot(
srt = ifnb_sub,
stat.by = c(
"db_qc", "outlier_qc", "umi_qc", "gene_qc",
"mito_qc", "ribo_qc", "ribo_mito_ratio_qc", "species_qc"
),
plot_type = "upset", stat_level = "Fail"
)
#> Warning: stat_type is forcibly set to 'count' when plot sankey, chord, venn or upset
table(pancreas_sub$CellQC)
#>
#> Pass Fail
#> 974 26
data("ifnb_sub")
ifnb_sub <- RunCellQC(ifnb_sub, split.by = "stim", UMI_threshold = 1000, gene_threshold = 550)
#> === CTRL ===
#> >>> Total cells: 1000
#> >>> Cells which are filtered out: 315
#> ... 54 potential doublets
#> ... 8 outlier cells
#> ... 28 low-UMI cells
#> ... 250 low-gene cells
#> ... 0 high-mito cells
#> ... 0 high-ribo cells
#> ... 0 ribo_mito_ratio outlier cells
#> ... 0 species-contaminated cells
#> >>> Remained cells after filtering: 685
#> === STIM ===
#> >>> Total cells: 1000
#> >>> Cells which are filtered out: 303
#> ... 42 potential doublets
#> ... 12 outlier cells
#> ... 25 low-UMI cells
#> ... 251 low-gene cells
#> ... 0 high-mito cells
#> ... 0 high-ribo cells
#> ... 0 ribo_mito_ratio outlier cells
#> ... 0 species-contaminated cells
#> >>> Remained cells after filtering: 697
CellStatPlot(
srt = ifnb_sub,
stat.by = c(
"db_qc", "outlier_qc", "umi_qc", "gene_qc",
"mito_qc", "ribo_qc", "ribo_mito_ratio_qc", "species_qc"
),
plot_type = "upset", stat_level = "Fail"
)
#> Warning: stat_type is forcibly set to 'count' when plot sankey, chord, venn or upset
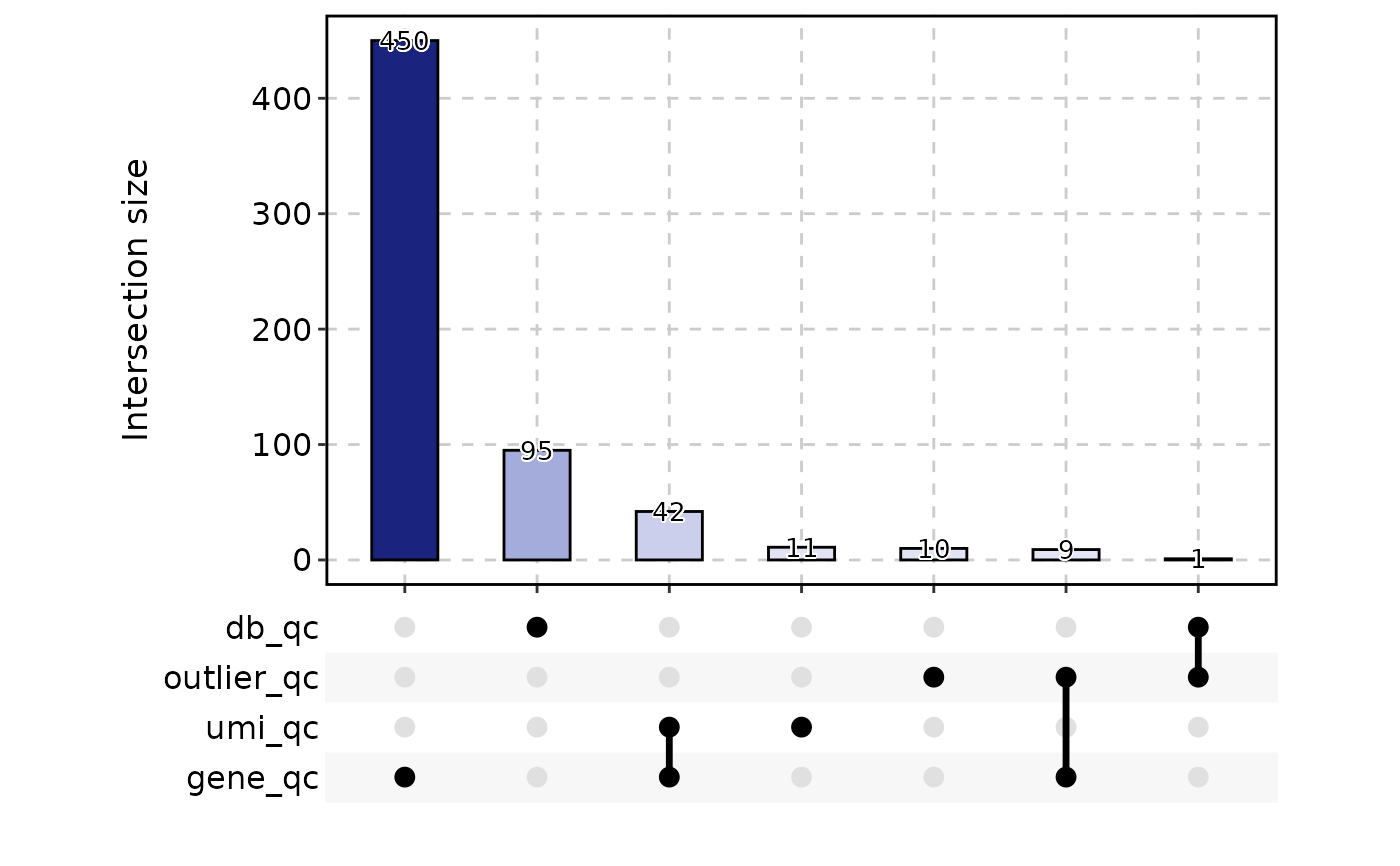 table(ifnb_sub$CellQC)
#>
#> Pass Fail
#> 1382 618
table(ifnb_sub$CellQC)
#>
#> Pass Fail
#> 1382 618