Identification of heterotypic (or neotypic) doublets in single-cell RNAseq data.
Usage
RunDoubletCalling(
srt,
assay = "RNA",
db_method = "scDblFinder",
db_rate = ncol(srt)/1000 * 0.01,
...
)Arguments
- srt
A Seurat object.
- assay
The name of the assay to be used for doublet-calling. Default is "RNA".
- db_method
Doublet-calling methods used. Can be one of
scDblFinder,Scrublet,DoubletDetection,scds_cxds,scds_bcds,scds_hybrid- db_rate
The expected doublet rate. By default this is assumed to be 1% per thousand cells captured (so 4% among 4000 thousand cells), which is appropriate for 10x datasets.
- ...
Arguments passed to the corresponding doublet-calling method.
Value
Returns Seurat object with the doublet prediction results and prediction scores stored in the meta.data slot.
Examples
data("pancreas_sub")
pancreas_sub <- RunDoubletCalling(pancreas_sub, db_method = "scDblFinder")
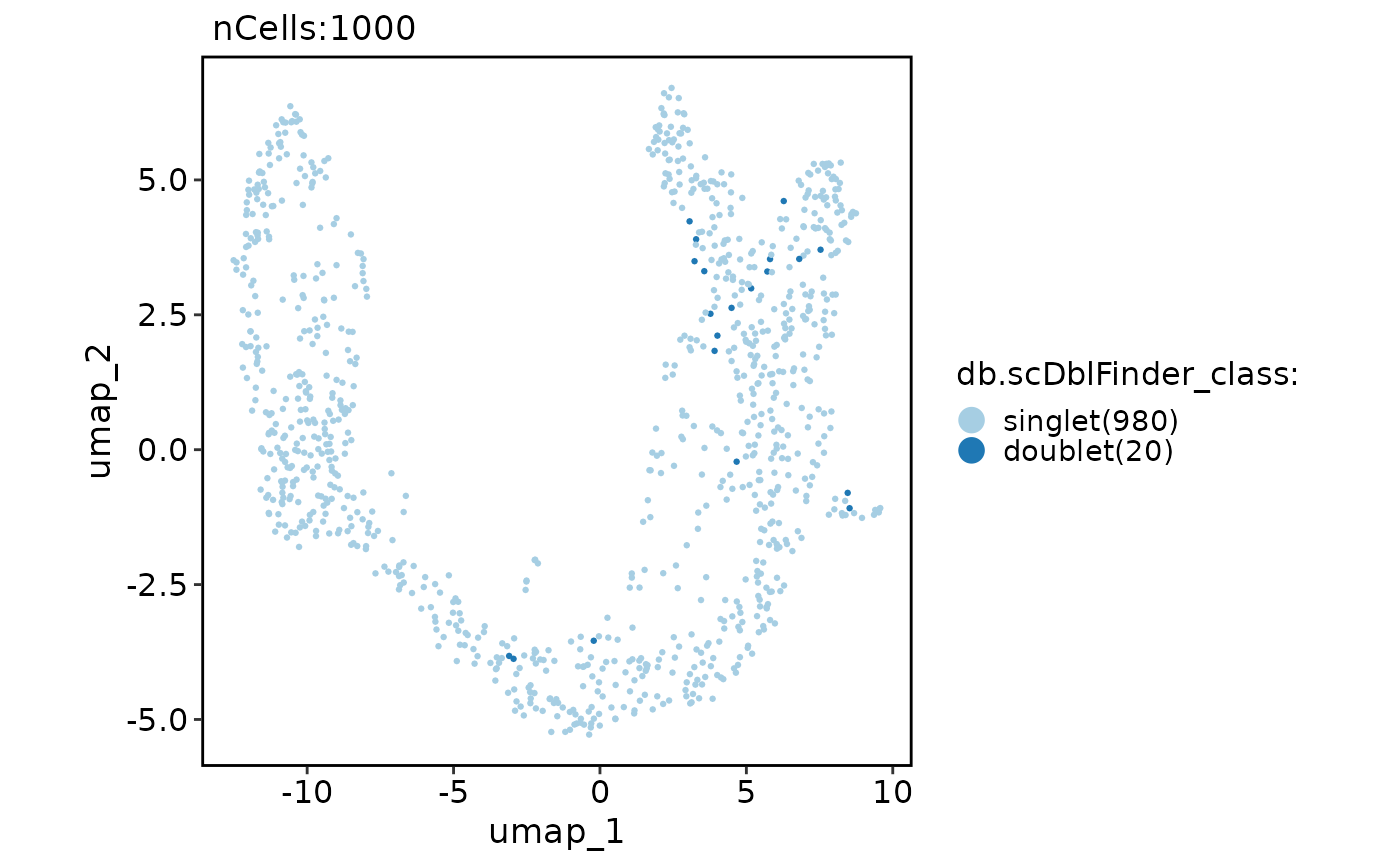
CellDimPlot(pancreas_sub, reduction = "umap", group.by = "db.scDblFinder_class")
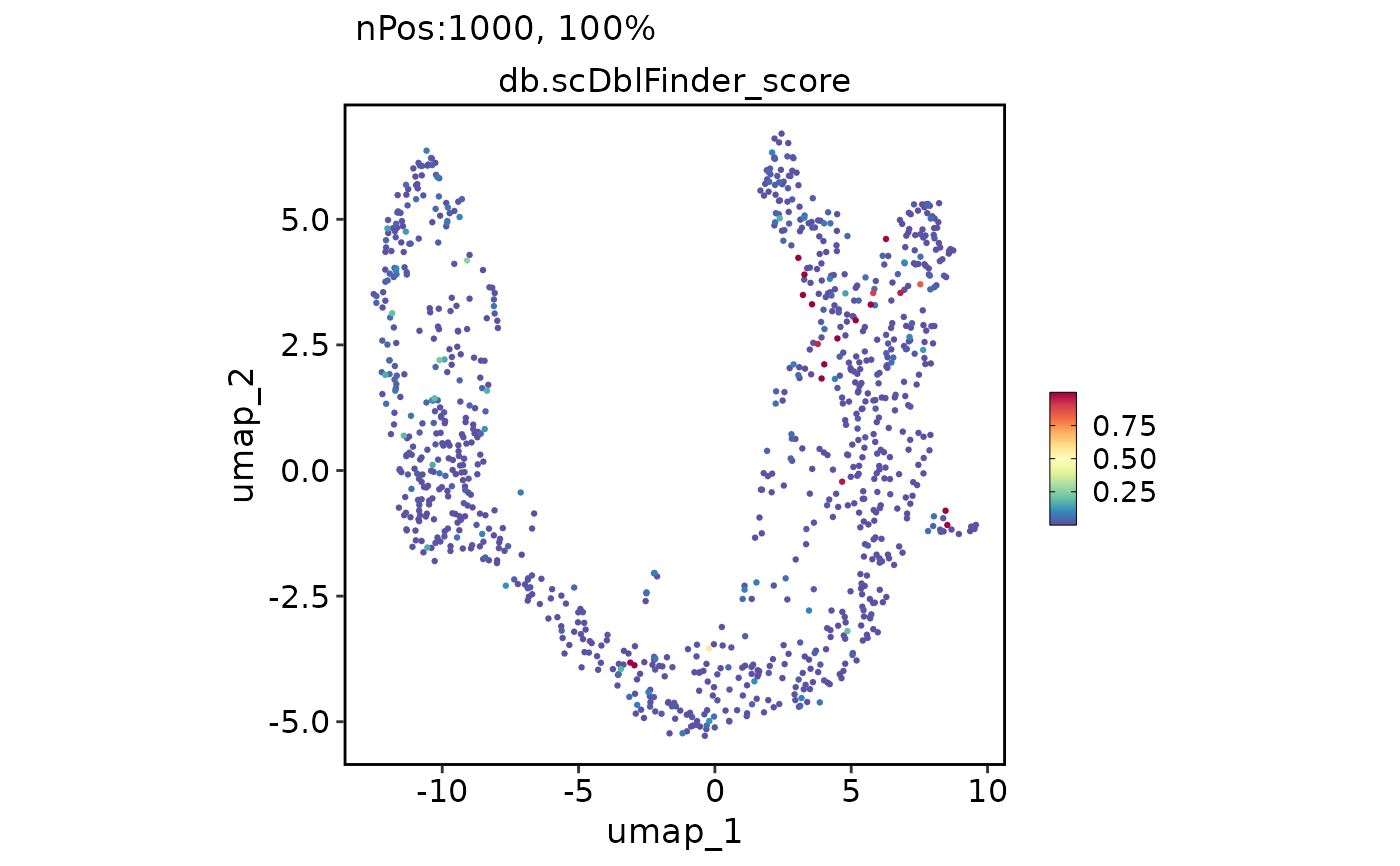
 FeatureDimPlot(pancreas_sub, reduction = "umap", features = "db.scDblFinder_score")
FeatureDimPlot(pancreas_sub, reduction = "umap", features = "db.scDblFinder_score")