Annotate single cells using scmap.
Usage
RunScmap(
srt_query,
srt_ref,
ref_group = NULL,
query_assay = "RNA",
ref_assay = "RNA",
method = "scmapCluster",
nfeatures = 500,
threshold = 0.5,
k = 10
)Arguments
- srt_query
An object of class Seurat to be annotated with cell types.
- srt_ref
An object of class Seurat storing the reference cells.
- ref_group
A character vector specifying the column name in the `srt_ref` metadata that represents the cell grouping.
- query_assay
A character vector specifying the assay to be used for the query data. Defaults to the default assay of the `srt_query` object.
- ref_assay
A character vector specifying the assay to be used for the reference data. Defaults to the default assay of the `srt_ref` object.
- method
The method to be used for scmap analysis. Can be any of "scmapCluster" or "scmapCell". The default value is "scmapCluster".
- nfeatures
The number of top features to be selected. The default value is 500.
- threshold
The threshold value on similarity to determine if a cell is assigned to a cluster. This should be a value between 0 and 1. The default value is 0.5.
- k
Number of clusters per group for k-means clustering when method is "scmapCell".
Examples
data("panc8_sub")
# Simply convert genes from human to mouse and preprocess the data
genenames <- make.unique(capitalize(rownames(panc8_sub), force_tolower = TRUE))
panc8_sub <- RenameFeatures(panc8_sub, newnames = genenames)
#> Rename features for the assay: RNA
panc8_sub <- check_srtMerge(panc8_sub, batch = "tech")[["srtMerge"]]
#> [2023-11-21 07:50:22.635964] Spliting srtMerge into srtList by column tech... ...
#> [2023-11-21 07:50:22.813549] Checking srtList... ...
#> Data 1/5 of the srtList is raw_normalized_counts. Perform NormalizeData(LogNormalize) on the data ...
#> Perform FindVariableFeatures on the data 1/5 of the srtList...
#> Data 2/5 of the srtList is raw_normalized_counts. Perform NormalizeData(LogNormalize) on the data ...
#> Perform FindVariableFeatures on the data 2/5 of the srtList...
#> Data 3/5 of the srtList is raw_normalized_counts. Perform NormalizeData(LogNormalize) on the data ...
#> Perform FindVariableFeatures on the data 3/5 of the srtList...
#> Data 4/5 of the srtList is raw_counts. Perform NormalizeData(LogNormalize) on the data ...
#> Perform FindVariableFeatures on the data 4/5 of the srtList...
#> Data 5/5 of the srtList is raw_counts. Perform NormalizeData(LogNormalize) on the data ...
#> Perform FindVariableFeatures on the data 5/5 of the srtList...
#> Use the separate HVF from srtList...
#> Number of available HVF: 2000
#> [2023-11-21 07:50:26.11454] Finished checking.
# Annotation
data("pancreas_sub")
pancreas_sub <- Standard_SCP(pancreas_sub)
#> [2023-11-21 07:50:27.795783] Start Standard_SCP
#> [2023-11-21 07:50:27.795974] Checking srtList... ...
#> Data 1/1 of the srtList is raw_counts. Perform NormalizeData(LogNormalize) on the data ...
#> Perform FindVariableFeatures on the data 1/1 of the srtList...
#> Use the separate HVF from srtList...
#> Number of available HVF: 2000
#> [2023-11-21 07:50:28.485316] Finished checking.
#> [2023-11-21 07:50:28.485507] Perform ScaleData on the data...
#> [2023-11-21 07:50:28.563784] Perform linear dimension reduction (pca) on the data...
#> Warning: The following arguments are not used: force.recalc
#> Warning: The following arguments are not used: force.recalc
#> [2023-11-21 07:50:29.137888] Perform FindClusters (louvain) on the data...
#> [2023-11-21 07:50:29.212299] Reorder clusters...
#> [2023-11-21 07:50:29.275877] Perform nonlinear dimension reduction (umap) on the data...
#> Non-linear dimensionality reduction(umap) using Reduction(Standardpca, dims:1-13) as input
#> Found more than one class "dist" in cache; using the first, from namespace 'BiocGenerics'
#> Also defined by ‘spam’
#> Found more than one class "dist" in cache; using the first, from namespace 'BiocGenerics'
#> Also defined by ‘spam’
#> Non-linear dimensionality reduction(umap) using Reduction(Standardpca, dims:1-13) as input
#> Found more than one class "dist" in cache; using the first, from namespace 'BiocGenerics'
#> Also defined by ‘spam’
#> Found more than one class "dist" in cache; using the first, from namespace 'BiocGenerics'
#> Also defined by ‘spam’
#> [2023-11-21 07:50:37.419439] Standard_SCP done
#> Elapsed time: 9.62 secs
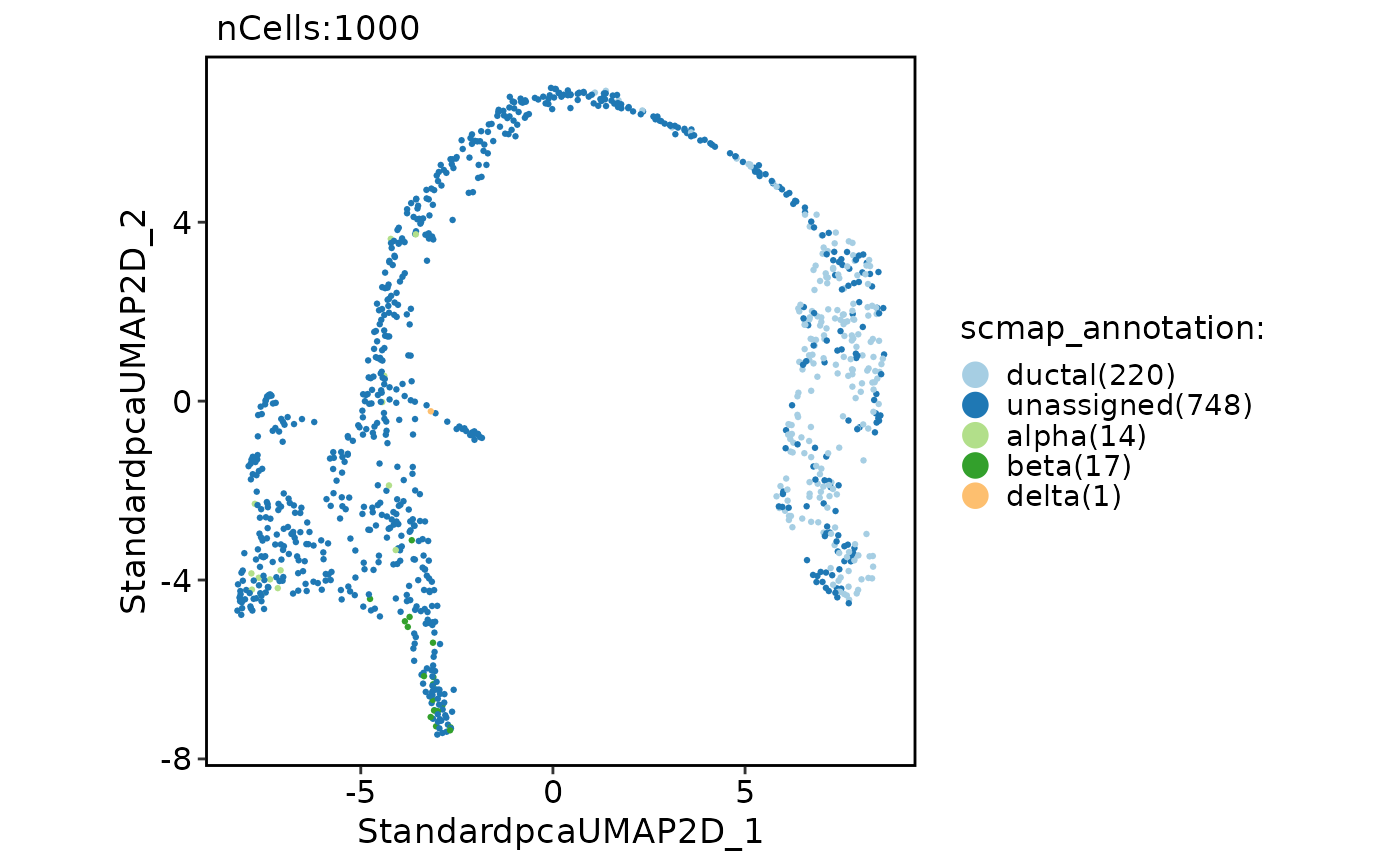
pancreas_sub <- RunScmap(
srt_query = pancreas_sub, srt_ref = panc8_sub,
ref_group = "celltype", method = "scmapCluster"
)
#> Detected srt_query data type: log_normalized_counts
#> Detected srt_ref data type: log_normalized_counts
#> Perform selectFeatures on the data...
#> Perform indexCluster on the data...
#> Perform scmapCluster on the data...
#> Warning: Features Adcyap1, Cfc1, Col6a2, Col6a3, Crp, Ctrc, Gsta1, Lcn2, Loxl4, Mafa, Mt-atp6, Mt-co1, Mt-co2, Mt-co3, Mt-nd1, Mt-nd2, Mt-nd4, Mt-nd4l, Mt-nd5, Muc13, Serpina5, Tacstd2 are not present in the 'SCESet' object and therefore were not set.
CellDimPlot(pancreas_sub, group.by = "scmap_annotation")
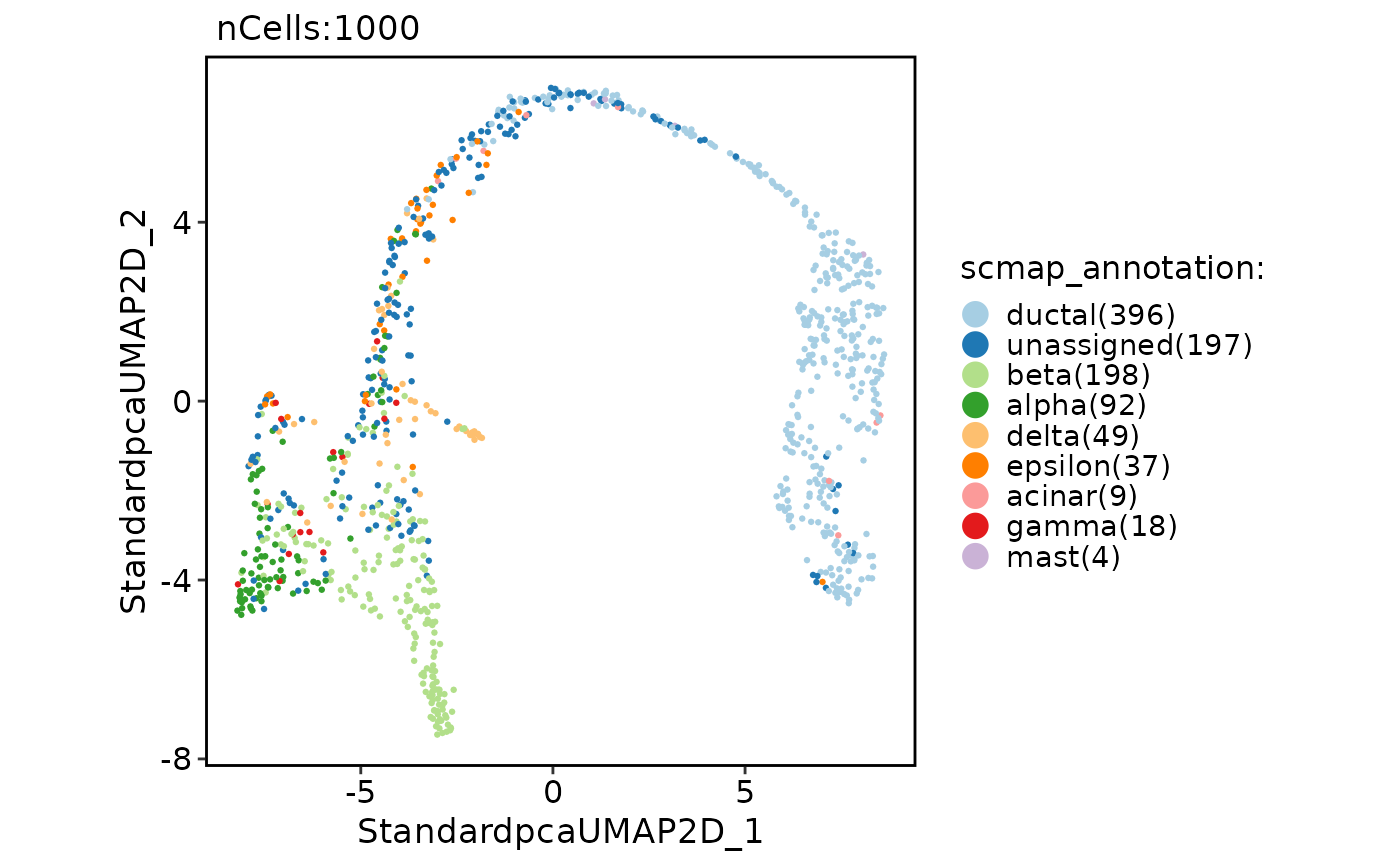 pancreas_sub <- RunScmap(
srt_query = pancreas_sub, srt_ref = panc8_sub,
ref_group = "celltype", method = "scmapCell"
)
#> Detected srt_query data type: log_normalized_counts
#> Detected srt_ref data type: log_normalized_counts
#> Perform selectFeatures on the data...
#> Perform indexCell on the data...
#> Perform scmapCell on the data...
#> Perform scmapCell2Cluster on the data...
CellDimPlot(pancreas_sub, group.by = "scmap_annotation")
pancreas_sub <- RunScmap(
srt_query = pancreas_sub, srt_ref = panc8_sub,
ref_group = "celltype", method = "scmapCell"
)
#> Detected srt_query data type: log_normalized_counts
#> Detected srt_ref data type: log_normalized_counts
#> Perform selectFeatures on the data...
#> Perform indexCell on the data...
#> Perform scmapCell on the data...
#> Perform scmapCell2Cluster on the data...
CellDimPlot(pancreas_sub, group.by = "scmap_annotation")