This function performs KNN prediction to annotate cell types based on reference scRNA-seq or bulk RNA-seq data.
Usage
RunKNNPredict(
srt_query,
srt_ref = NULL,
bulk_ref = NULL,
query_group = NULL,
ref_group = NULL,
query_assay = NULL,
ref_assay = NULL,
query_reduction = NULL,
ref_reduction = NULL,
query_dims = 1:30,
ref_dims = 1:30,
query_collapsing = !is.null(query_group),
ref_collapsing = TRUE,
return_full_distance_matrix = FALSE,
features = NULL,
features_type = c("HVF", "DE"),
feature_source = "both",
nfeatures = 2000,
DEtest_param = list(max.cells.per.ident = 200, test.use = "wilcox"),
DE_threshold = "p_val_adj < 0.05",
nn_method = NULL,
distance_metric = "cosine",
k = 30,
filter_lowfreq = 0,
prefix = "KNNPredict"
)Arguments
- srt_query
An object of class Seurat to be annotated with cell types.
- srt_ref
An object of class Seurat storing the reference cells.
- bulk_ref
A cell atlas matrix, where cell types are represented by columns and genes are represented by rows, for example, SCP::ref_scHCL. Either `srt_ref` or `bulk_ref` must be provided.
- query_group
A character vector specifying the column name in the `srt_query` metadata that represents the cell grouping.
- ref_group
A character vector specifying the column name in the `srt_ref` metadata that represents the cell grouping.
- query_assay
A character vector specifying the assay to be used for the query data. Defaults to the default assay of the `srt_query` object.
- ref_assay
A character vector specifying the assay to be used for the reference data. Defaults to the default assay of the `srt_ref` object.
- query_reduction
A character vector specifying the dimensionality reduction method used for the query data. If NULL, the function will use the default reduction method specified in the `srt_query` object.
- ref_reduction
A character vector specifying the dimensionality reduction method used for the reference data. If NULL, the function will use the default reduction method specified in the `srt_ref` object.
- query_dims
A numeric vector specifying the dimensions to be used for the query data. Defaults to the first 30 dimensions.
- ref_dims
A numeric vector specifying the dimensions to be used for the reference data. Defaults to the first 30 dimensions.
- query_collapsing
A boolean value indicating whether the query data should be collapsed to group-level average expression values. If TRUE, the function will calculate the average expression values for each group in the query data and the annotation will be performed separately for each group. Otherwise it will use the raw expression values for each cell.
- ref_collapsing
A boolean value indicating whether the reference data should be collapsed to group-level average expression values. If TRUE, the function will calculate the average expression values for each group in the reference data and the annotation will be performed separately for each group. Otherwise it will use the raw expression values for each cell.
- return_full_distance_matrix
A boolean value indicating whether the full distance matrix should be returned. If TRUE, the function will return the distance matrix used for the KNN prediction, otherwise it will only return the annotated cell types.
- features
A character vector specifying the features (genes) to be used for the KNN prediction. If NULL, all the features in the query and reference data will be used.
- features_type
A character vector specifying the type of features to be used for the KNN prediction. Must be one of "HVF" (highly variable features) or "DE" (differentially expressed features). Defaults to "HVF".
- feature_source
A character vector specifying the source of the features to be used for the KNN prediction. Must be one of "both", "query", or "ref". Defaults to "both".
- nfeatures
An integer specifying the maximum number of features to be used for the KNN prediction. Defaults to 2000.
- DEtest_param
A list of parameters to be passed to the differential expression test function if `features_type` is set to "DE". Defaults to `list(max.cells.per.ident = 200, test.use = "wilcox")`.
- DE_threshold
Threshold used to filter the DE features. Default is "p_val < 0.05". If using "roc" test,
DE_thresholdshould be needs to be reassigned. e.g. "power > 0.5".- nn_method
A character vector specifying the method to be used for finding nearest neighbors. Must be one of "raw", "rann", or "annoy". Defaults to "raw".
- distance_metric
A character vector specifying the distance metric to be used for calculating similarity between cells. Must be one of "cosine", "euclidean", "manhattan", or "hamming". Defaults to "cosine".
- k
An integer specifying the number of nearest neighbors to be considered for the KNN prediction. Defaults to 30.
- filter_lowfreq
An integer specifying the threshold for filtering low-frequency cell types from the predicted results. Cell types with a frequency lower than `filter_lowfreq` will be labelled as "unreliable". Defaults to 0, which means no filtering will be performed.
- prefix
A character vector specifying the prefix to be added to the resulting annotations. Defaults to "KNNPredict".
Examples
# Annotate cells using bulk RNA-seq data
data("pancreas_sub")
data("ref_scMCA")
pancreas_sub <- Standard_SCP(pancreas_sub)
#> [2023-11-21 07:43:25.690838] Start Standard_SCP
#> [2023-11-21 07:43:25.691023] Checking srtList... ...
#> Data 1/1 of the srtList is raw_counts. Perform NormalizeData(LogNormalize) on the data ...
#> Perform FindVariableFeatures on the data 1/1 of the srtList...
#> Use the separate HVF from srtList...
#> Number of available HVF: 2000
#> [2023-11-21 07:43:26.39611] Finished checking.
#> [2023-11-21 07:43:26.396329] Perform ScaleData on the data...
#> [2023-11-21 07:43:26.47502] Perform linear dimension reduction (pca) on the data...
#> Warning: The following arguments are not used: force.recalc
#> Warning: The following arguments are not used: force.recalc
#> [2023-11-21 07:43:27.052901] Perform FindClusters (louvain) on the data...
#> [2023-11-21 07:43:27.127977] Reorder clusters...
#> [2023-11-21 07:43:27.190403] Perform nonlinear dimension reduction (umap) on the data...
#> Non-linear dimensionality reduction(umap) using Reduction(Standardpca, dims:1-13) as input
#> Found more than one class "dist" in cache; using the first, from namespace 'BiocGenerics'
#> Also defined by ‘spam’
#> Found more than one class "dist" in cache; using the first, from namespace 'BiocGenerics'
#> Also defined by ‘spam’
#> Non-linear dimensionality reduction(umap) using Reduction(Standardpca, dims:1-13) as input
#> Found more than one class "dist" in cache; using the first, from namespace 'BiocGenerics'
#> Also defined by ‘spam’
#> Found more than one class "dist" in cache; using the first, from namespace 'BiocGenerics'
#> Also defined by ‘spam’
#> [2023-11-21 07:43:35.254245] Standard_SCP done
#> Elapsed time: 9.56 secs
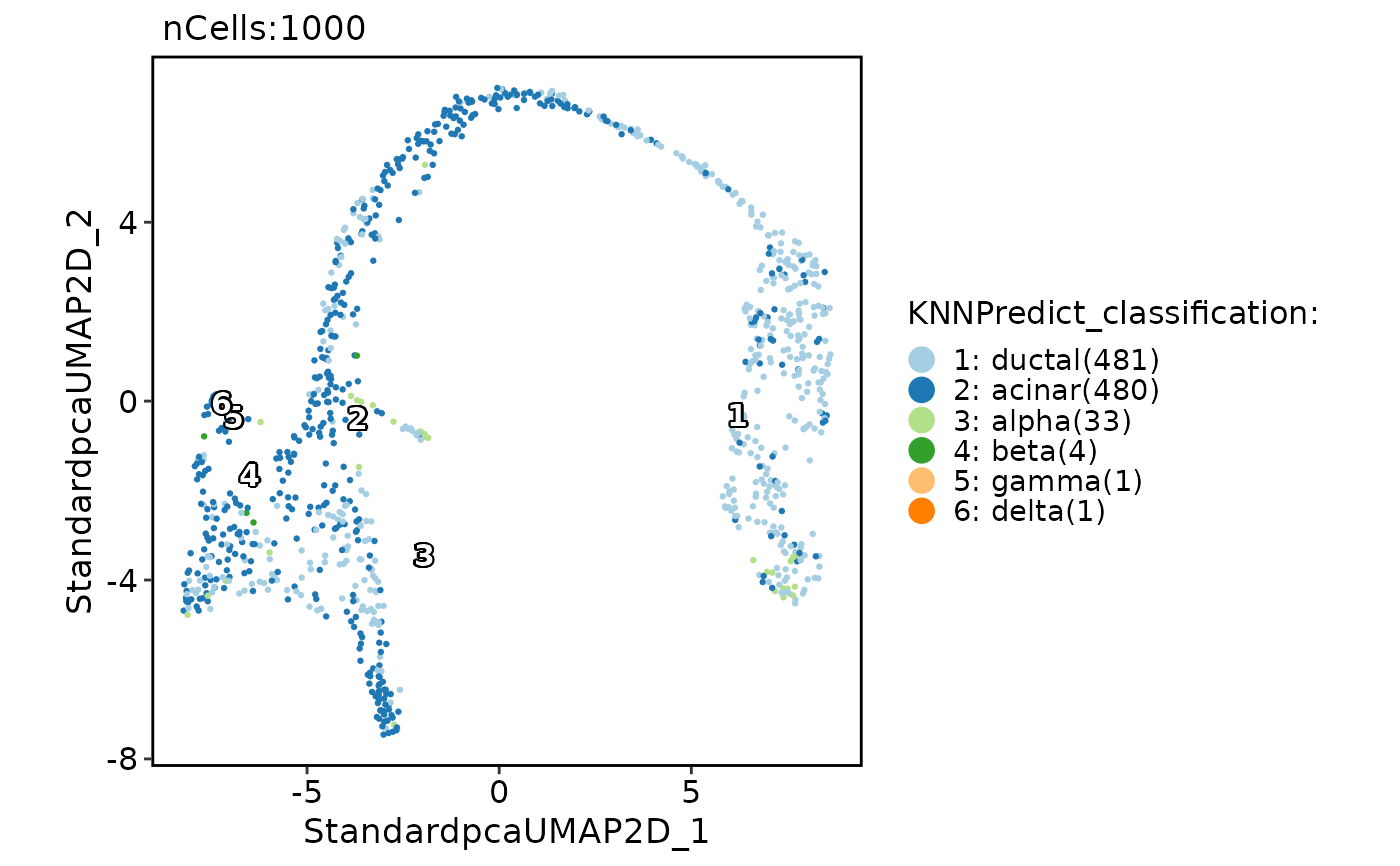
pancreas_sub <- RunKNNPredict(srt_query = pancreas_sub, bulk_ref = ref_scMCA)
#> Use 535 features to calculate distance.
#> Detected query data type: log_normalized_counts
#> Detected reference data type: log_normalized_counts
#> Calculate similarity...
#> Use 'raw' method to find neighbors.
#> Predict cell type...
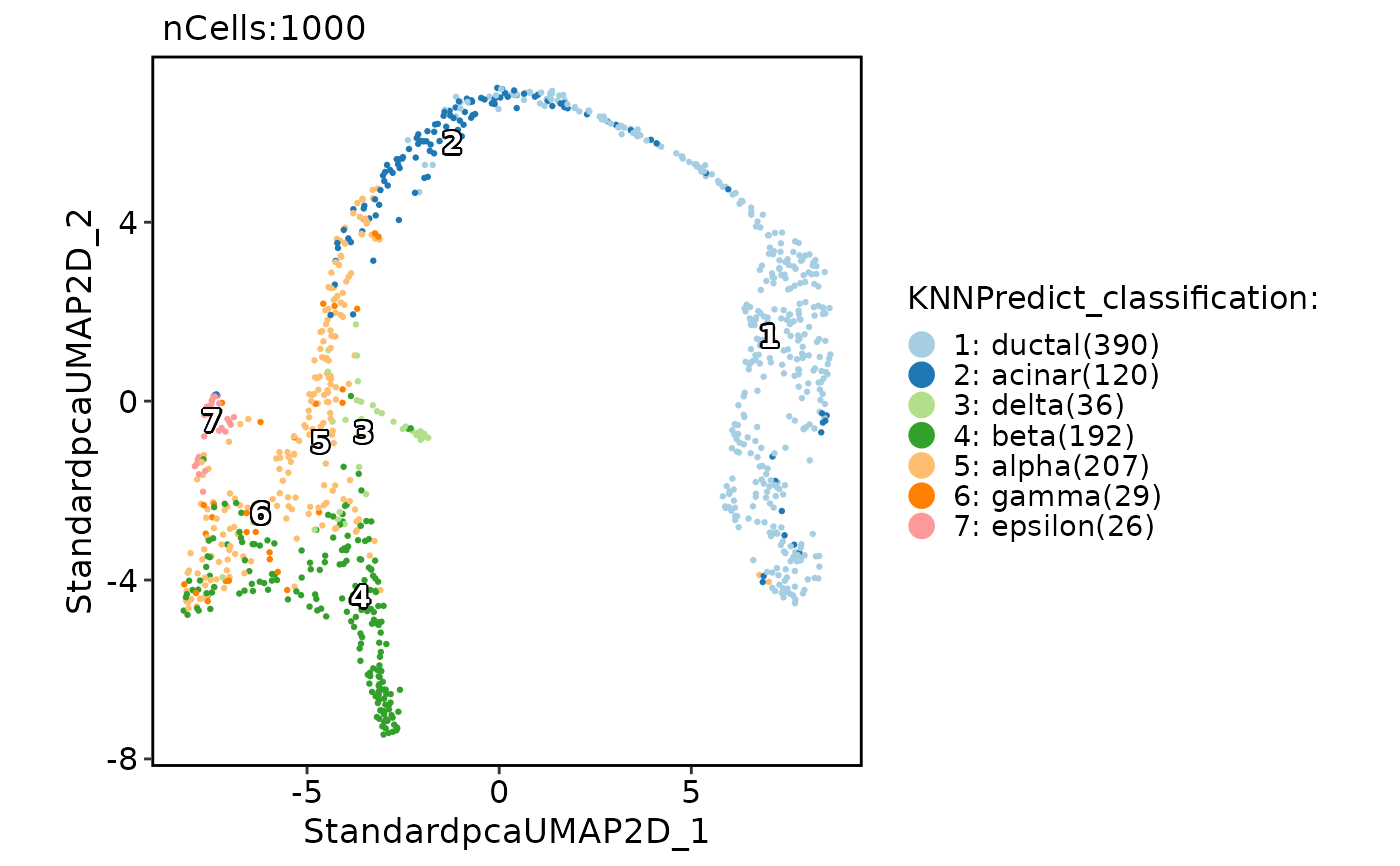
CellDimPlot(pancreas_sub, group.by = "KNNPredict_classification", label = TRUE)
 # Removal of low credible cell types from the predicted results
pancreas_sub <- RunKNNPredict(srt_query = pancreas_sub, bulk_ref = ref_scMCA, filter_lowfreq = 30)
#> Use 535 features to calculate distance.
#> Detected query data type: log_normalized_counts
#> Detected reference data type: log_normalized_counts
#> Calculate similarity...
#> Use 'raw' method to find neighbors.
#> Predict cell type...
CellDimPlot(pancreas_sub, group.by = "KNNPredict_classification", label = TRUE)
# Removal of low credible cell types from the predicted results
pancreas_sub <- RunKNNPredict(srt_query = pancreas_sub, bulk_ref = ref_scMCA, filter_lowfreq = 30)
#> Use 535 features to calculate distance.
#> Detected query data type: log_normalized_counts
#> Detected reference data type: log_normalized_counts
#> Calculate similarity...
#> Use 'raw' method to find neighbors.
#> Predict cell type...
CellDimPlot(pancreas_sub, group.by = "KNNPredict_classification", label = TRUE)
 # Annotate clusters using bulk RNA-seq data
pancreas_sub <- RunKNNPredict(srt_query = pancreas_sub, query_group = "SubCellType", bulk_ref = ref_scMCA)
#> Use 535 features to calculate distance.
#> Detected query data type: log_normalized_counts
#> Detected reference data type: log_normalized_counts
#> Calculate similarity...
#> Use 'raw' method to find neighbors.
#> Predict cell type...
#> Error: No cell overlap between new meta data and Seurat object
CellDimPlot(pancreas_sub, group.by = "KNNPredict_classification", label = TRUE)
# Annotate clusters using bulk RNA-seq data
pancreas_sub <- RunKNNPredict(srt_query = pancreas_sub, query_group = "SubCellType", bulk_ref = ref_scMCA)
#> Use 535 features to calculate distance.
#> Detected query data type: log_normalized_counts
#> Detected reference data type: log_normalized_counts
#> Calculate similarity...
#> Use 'raw' method to find neighbors.
#> Predict cell type...
#> Error: No cell overlap between new meta data and Seurat object
CellDimPlot(pancreas_sub, group.by = "KNNPredict_classification", label = TRUE)
 # Annotate using single cell RNA-seq data
data("panc8_sub")
# Simply convert genes from human to mouse and preprocess the data
genenames <- make.unique(capitalize(rownames(panc8_sub), force_tolower = TRUE))
panc8_sub <- RenameFeatures(panc8_sub, newnames = genenames)
#> Rename features for the assay: RNA
panc8_sub <- check_srtMerge(panc8_sub, batch = "tech")[["srtMerge"]]
#> [2023-11-21 07:43:37.918638] Spliting srtMerge into srtList by column tech... ...
#> [2023-11-21 07:43:38.077584] Checking srtList... ...
#> Data 1/5 of the srtList is raw_normalized_counts. Perform NormalizeData(LogNormalize) on the data ...
#> Perform FindVariableFeatures on the data 1/5 of the srtList...
#> Data 2/5 of the srtList is raw_normalized_counts. Perform NormalizeData(LogNormalize) on the data ...
#> Perform FindVariableFeatures on the data 2/5 of the srtList...
#> Data 3/5 of the srtList is raw_normalized_counts. Perform NormalizeData(LogNormalize) on the data ...
#> Perform FindVariableFeatures on the data 3/5 of the srtList...
#> Data 4/5 of the srtList is raw_counts. Perform NormalizeData(LogNormalize) on the data ...
#> Perform FindVariableFeatures on the data 4/5 of the srtList...
#> Data 5/5 of the srtList is raw_counts. Perform NormalizeData(LogNormalize) on the data ...
#> Perform FindVariableFeatures on the data 5/5 of the srtList...
#> Use the separate HVF from srtList...
#> Number of available HVF: 2000
#> [2023-11-21 07:43:39.867239] Finished checking.
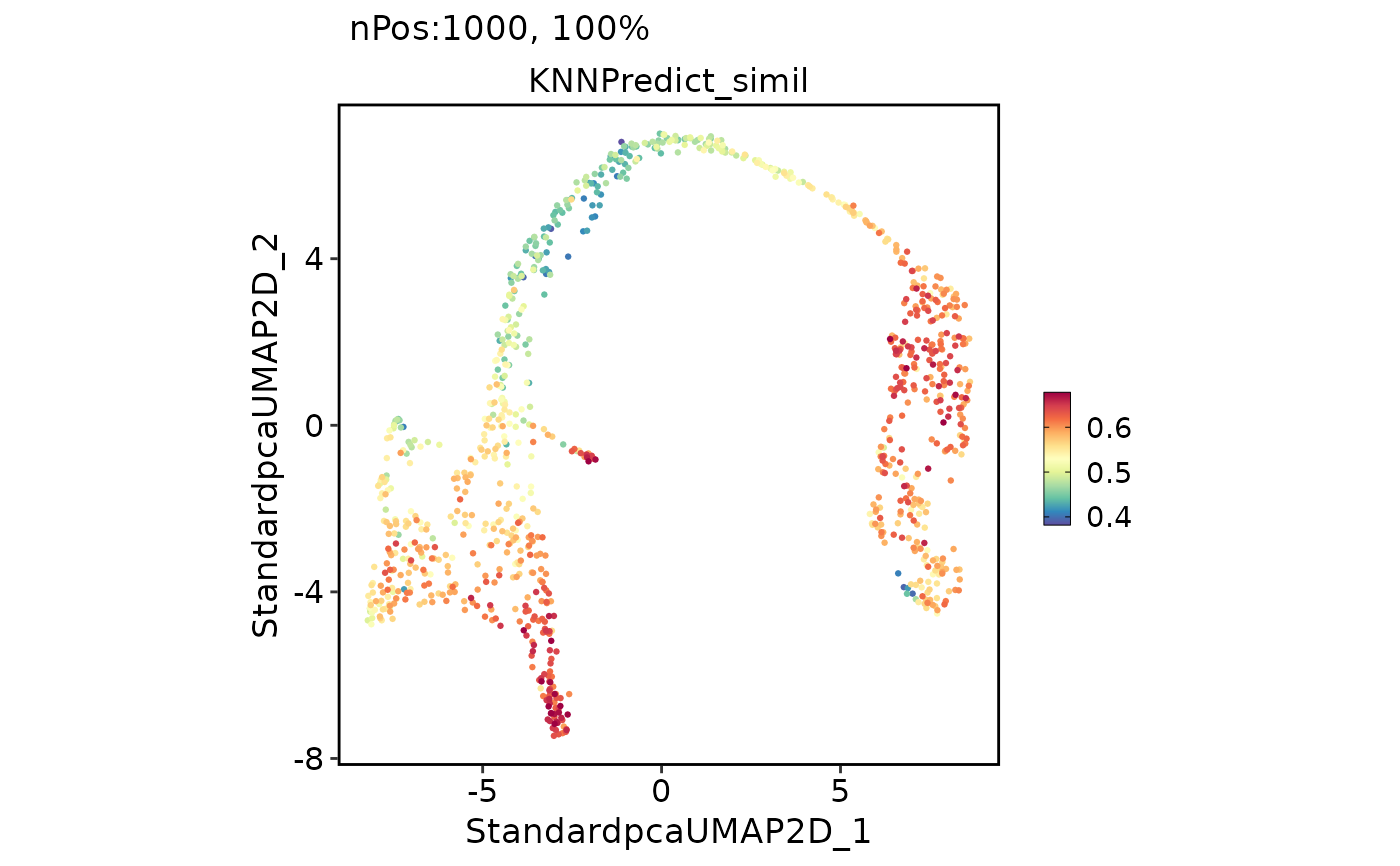
pancreas_sub <- RunKNNPredict(srt_query = pancreas_sub, srt_ref = panc8_sub, ref_group = "celltype")
#> Use the HVF to calculate distance metric.
#> Use 631 features to calculate distance.
#> Detected query data type: log_normalized_counts
#> Detected reference data type: log_normalized_counts
#> Calculate similarity...
#> Use 'raw' method to find neighbors.
#> Predict cell type...
CellDimPlot(pancreas_sub, group.by = "KNNPredict_classification", label = TRUE)
# Annotate using single cell RNA-seq data
data("panc8_sub")
# Simply convert genes from human to mouse and preprocess the data
genenames <- make.unique(capitalize(rownames(panc8_sub), force_tolower = TRUE))
panc8_sub <- RenameFeatures(panc8_sub, newnames = genenames)
#> Rename features for the assay: RNA
panc8_sub <- check_srtMerge(panc8_sub, batch = "tech")[["srtMerge"]]
#> [2023-11-21 07:43:37.918638] Spliting srtMerge into srtList by column tech... ...
#> [2023-11-21 07:43:38.077584] Checking srtList... ...
#> Data 1/5 of the srtList is raw_normalized_counts. Perform NormalizeData(LogNormalize) on the data ...
#> Perform FindVariableFeatures on the data 1/5 of the srtList...
#> Data 2/5 of the srtList is raw_normalized_counts. Perform NormalizeData(LogNormalize) on the data ...
#> Perform FindVariableFeatures on the data 2/5 of the srtList...
#> Data 3/5 of the srtList is raw_normalized_counts. Perform NormalizeData(LogNormalize) on the data ...
#> Perform FindVariableFeatures on the data 3/5 of the srtList...
#> Data 4/5 of the srtList is raw_counts. Perform NormalizeData(LogNormalize) on the data ...
#> Perform FindVariableFeatures on the data 4/5 of the srtList...
#> Data 5/5 of the srtList is raw_counts. Perform NormalizeData(LogNormalize) on the data ...
#> Perform FindVariableFeatures on the data 5/5 of the srtList...
#> Use the separate HVF from srtList...
#> Number of available HVF: 2000
#> [2023-11-21 07:43:39.867239] Finished checking.
pancreas_sub <- RunKNNPredict(srt_query = pancreas_sub, srt_ref = panc8_sub, ref_group = "celltype")
#> Use the HVF to calculate distance metric.
#> Use 631 features to calculate distance.
#> Detected query data type: log_normalized_counts
#> Detected reference data type: log_normalized_counts
#> Calculate similarity...
#> Use 'raw' method to find neighbors.
#> Predict cell type...
CellDimPlot(pancreas_sub, group.by = "KNNPredict_classification", label = TRUE)
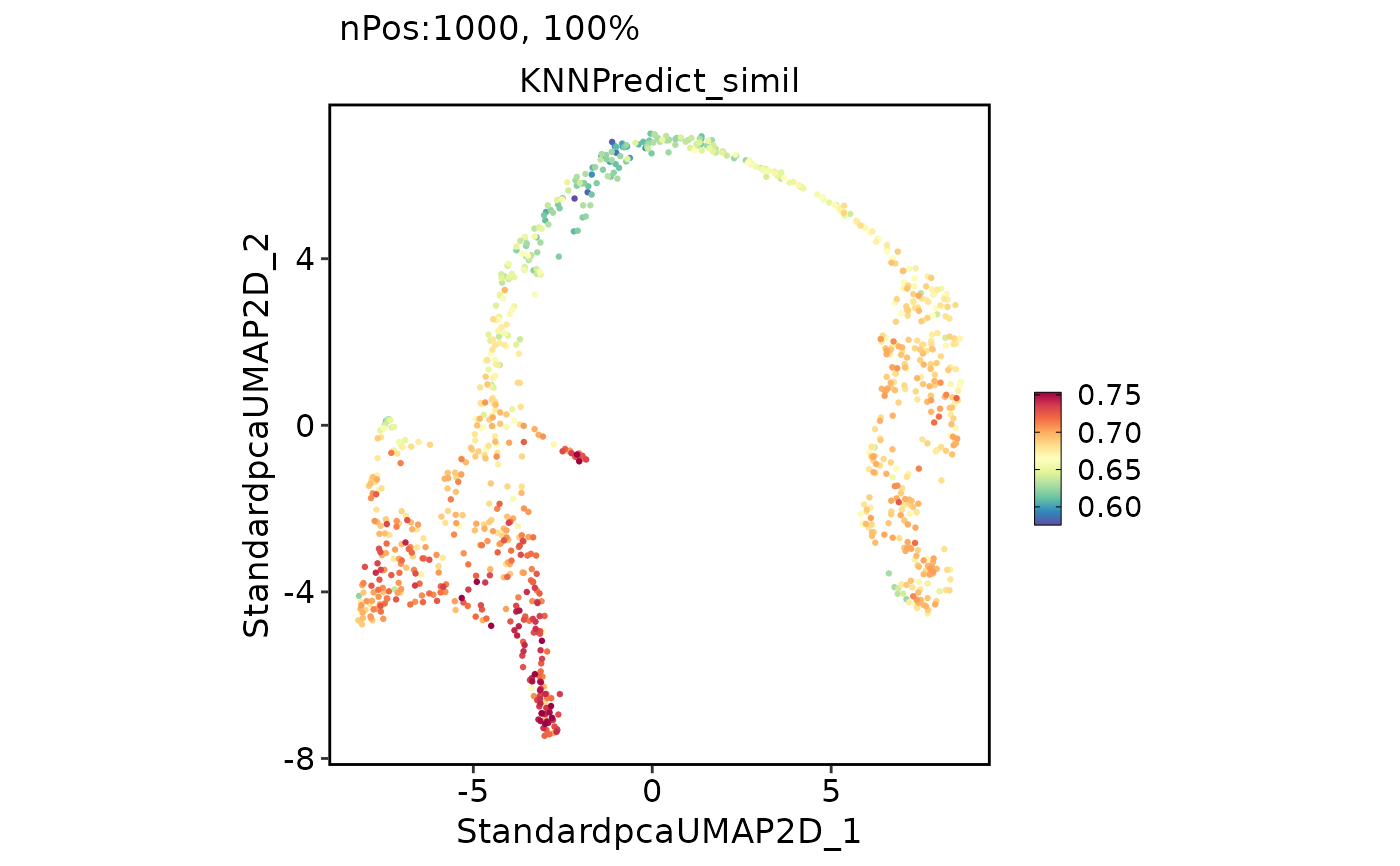
 FeatureDimPlot(pancreas_sub, features = "KNNPredict_simil")
FeatureDimPlot(pancreas_sub, features = "KNNPredict_simil")
 pancreas_sub <- RunKNNPredict(
srt_query = pancreas_sub, srt_ref = panc8_sub,
ref_group = "celltype", ref_collapsing = FALSE
)
#> Use the HVF to calculate distance metric.
#> Use 631 features to calculate distance.
#> Detected query data type: log_normalized_counts
#> Detected reference data type: log_normalized_counts
#> Calculate similarity...
#> Use 'raw' method to find neighbors.
#> Predict cell type...
CellDimPlot(pancreas_sub, group.by = "KNNPredict_classification", label = TRUE)
pancreas_sub <- RunKNNPredict(
srt_query = pancreas_sub, srt_ref = panc8_sub,
ref_group = "celltype", ref_collapsing = FALSE
)
#> Use the HVF to calculate distance metric.
#> Use 631 features to calculate distance.
#> Detected query data type: log_normalized_counts
#> Detected reference data type: log_normalized_counts
#> Calculate similarity...
#> Use 'raw' method to find neighbors.
#> Predict cell type...
CellDimPlot(pancreas_sub, group.by = "KNNPredict_classification", label = TRUE)
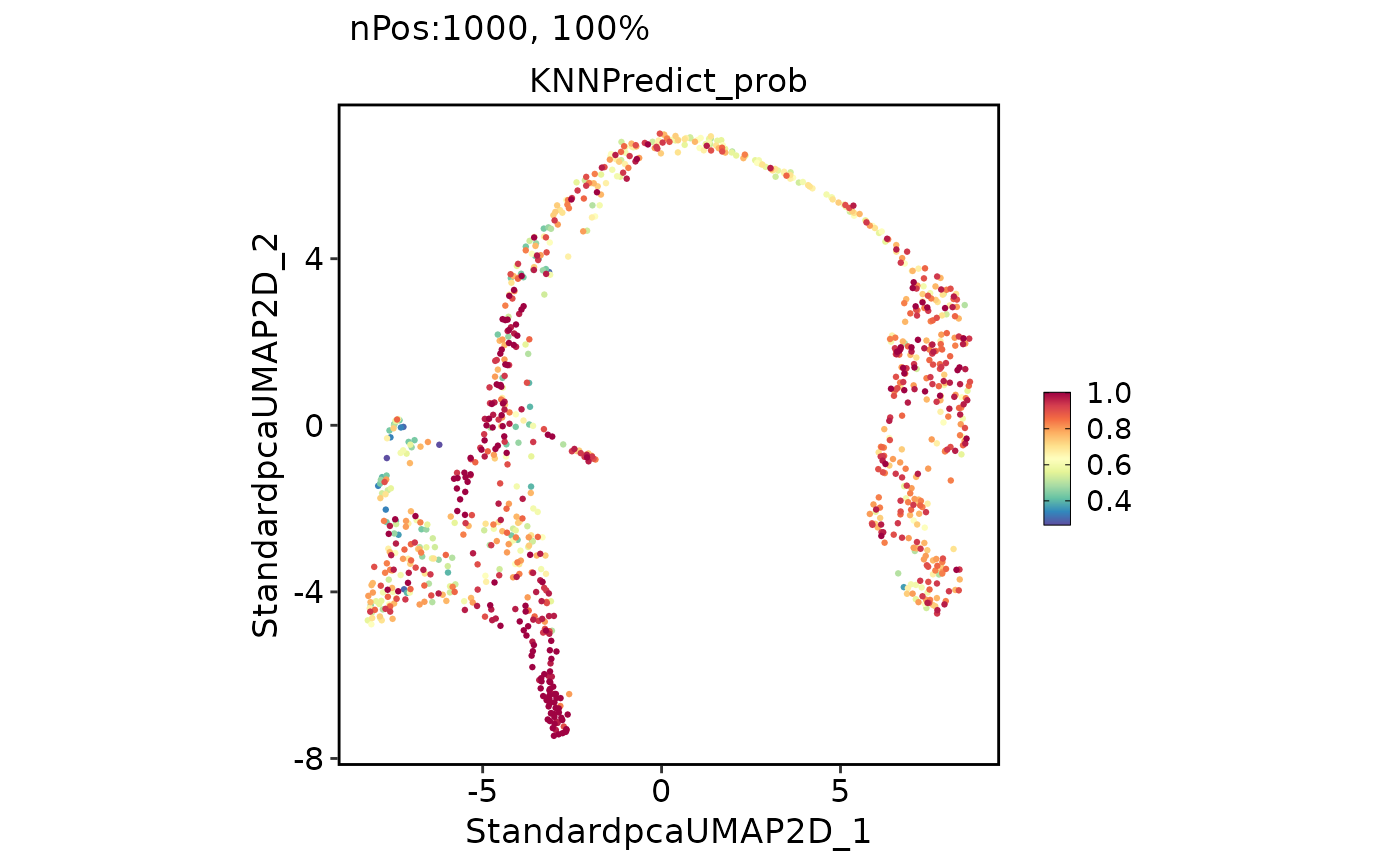
 FeatureDimPlot(pancreas_sub, features = "KNNPredict_prob")
FeatureDimPlot(pancreas_sub, features = "KNNPredict_prob")
 pancreas_sub <- RunKNNPredict(
srt_query = pancreas_sub, srt_ref = panc8_sub,
query_group = "SubCellType", ref_group = "celltype"
)
#> Use the HVF to calculate distance metric.
#> Use 631 features to calculate distance.
#> Detected query data type: log_normalized_counts
#> Detected reference data type: log_normalized_counts
#> Calculate similarity...
#> Use 'raw' method to find neighbors.
#> Predict cell type...
#> Error: No cell overlap between new meta data and Seurat object
CellDimPlot(pancreas_sub, group.by = "KNNPredict_classification", label = TRUE)
pancreas_sub <- RunKNNPredict(
srt_query = pancreas_sub, srt_ref = panc8_sub,
query_group = "SubCellType", ref_group = "celltype"
)
#> Use the HVF to calculate distance metric.
#> Use 631 features to calculate distance.
#> Detected query data type: log_normalized_counts
#> Detected reference data type: log_normalized_counts
#> Calculate similarity...
#> Use 'raw' method to find neighbors.
#> Predict cell type...
#> Error: No cell overlap between new meta data and Seurat object
CellDimPlot(pancreas_sub, group.by = "KNNPredict_classification", label = TRUE)
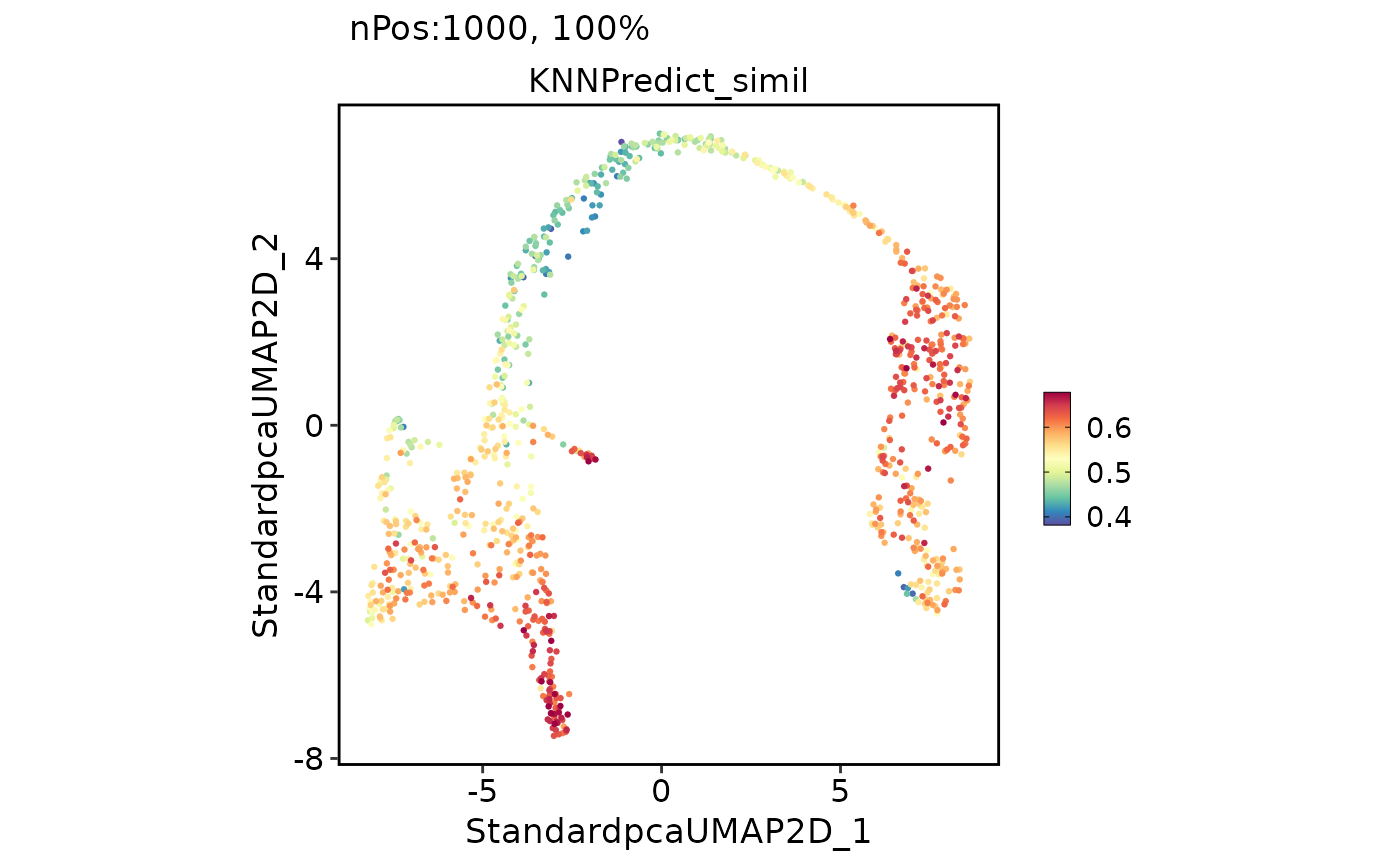
 FeatureDimPlot(pancreas_sub, features = "KNNPredict_simil")
FeatureDimPlot(pancreas_sub, features = "KNNPredict_simil")
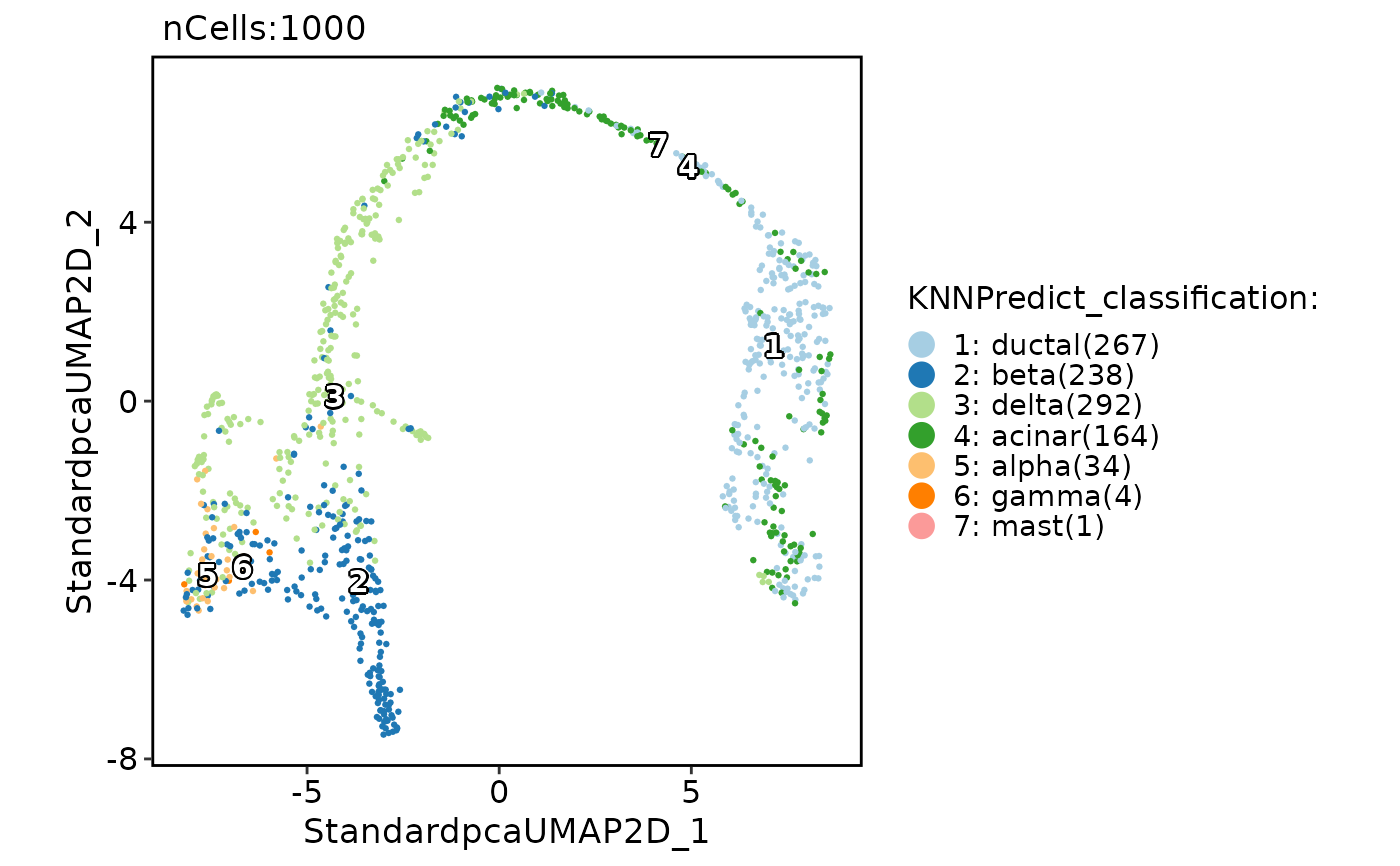
 # Annotate with DE gene instead of HVF
pancreas_sub <- RunKNNPredict(
srt_query = pancreas_sub, srt_ref = panc8_sub,
ref_group = "celltype",
features_type = "DE", feature_source = "ref"
)
#> [2023-11-21 07:43:42.979116] Start DEtest
#> Workers: 2
#> Find all markers(wilcox) among 13 groups...
#>
|
| | 0%
#>
#>
#>
#>
#>
#>
#>
#>
|
|=========================================================== | 54%
#>
#>
#>
#>
#>
#>
|
|==============================================================================================================| 100%
#>
#> [2023-11-21 07:44:07.47174] DEtest done
#> Elapsed time:24.49 secs
#> Use the DE features from AllMarkers_wilcox to calculate distance metric.
#> DE features number of the ref data: 2000
#> Use 1812 features to calculate distance.
#> Detected query data type: log_normalized_counts
#> Detected reference data type: log_normalized_counts
#> Calculate similarity...
#> Use 'raw' method to find neighbors.
#> Predict cell type...
CellDimPlot(pancreas_sub, group.by = "KNNPredict_classification", label = TRUE)
# Annotate with DE gene instead of HVF
pancreas_sub <- RunKNNPredict(
srt_query = pancreas_sub, srt_ref = panc8_sub,
ref_group = "celltype",
features_type = "DE", feature_source = "ref"
)
#> [2023-11-21 07:43:42.979116] Start DEtest
#> Workers: 2
#> Find all markers(wilcox) among 13 groups...
#>
|
| | 0%
#>
#>
#>
#>
#>
#>
#>
#>
|
|=========================================================== | 54%
#>
#>
#>
#>
#>
#>
|
|==============================================================================================================| 100%
#>
#> [2023-11-21 07:44:07.47174] DEtest done
#> Elapsed time:24.49 secs
#> Use the DE features from AllMarkers_wilcox to calculate distance metric.
#> DE features number of the ref data: 2000
#> Use 1812 features to calculate distance.
#> Detected query data type: log_normalized_counts
#> Detected reference data type: log_normalized_counts
#> Calculate similarity...
#> Use 'raw' method to find neighbors.
#> Predict cell type...
CellDimPlot(pancreas_sub, group.by = "KNNPredict_classification", label = TRUE)
 FeatureDimPlot(pancreas_sub, features = "KNNPredict_simil")
FeatureDimPlot(pancreas_sub, features = "KNNPredict_simil")
 pancreas_sub <- RunKNNPredict(
srt_query = pancreas_sub, srt_ref = panc8_sub,
query_group = "SubCellType", ref_group = "celltype",
features_type = "DE", feature_source = "both"
)
#> [2023-11-21 07:44:08.264926] Start DEtest
#> Workers: 2
#> Find all markers(wilcox) among 8 groups...
#>
|
| | 0%
#>
#>
#>
#>
#>
|
|======================================================= | 50%
#>
#>
#>
#>
#>
|
|==============================================================================================================| 100%
#>
#> [2023-11-21 07:44:15.497146] DEtest done
#> Elapsed time:7.23 secs
#> Use the DE features from AllMarkers_wilcox to calculate distance metric.
#> DE features number of the query data: 2000
#> [2023-11-21 07:44:15.795602] Start DEtest
#> Workers: 2
#> Find all markers(wilcox) among 13 groups...
#>
|
| | 0%
#>
#>
#>
#>
#>
#>
#>
#>
|
|=========================================================== | 54%
#>
#>
#>
#>
#>
#>
|
|==============================================================================================================| 100%
#>
#> [2023-11-21 07:44:40.172202] DEtest done
#> Elapsed time:24.38 secs
#> Use the DE features from AllMarkers_wilcox to calculate distance metric.
#> DE features number of the ref data: 650
#> Use 181 features to calculate distance.
#> Detected query data type: log_normalized_counts
#> Detected reference data type: log_normalized_counts
#> Calculate similarity...
#> Use 'raw' method to find neighbors.
#> Predict cell type...
#> Error: No cell overlap between new meta data and Seurat object
CellDimPlot(pancreas_sub, group.by = "KNNPredict_classification", label = TRUE)
pancreas_sub <- RunKNNPredict(
srt_query = pancreas_sub, srt_ref = panc8_sub,
query_group = "SubCellType", ref_group = "celltype",
features_type = "DE", feature_source = "both"
)
#> [2023-11-21 07:44:08.264926] Start DEtest
#> Workers: 2
#> Find all markers(wilcox) among 8 groups...
#>
|
| | 0%
#>
#>
#>
#>
#>
|
|======================================================= | 50%
#>
#>
#>
#>
#>
|
|==============================================================================================================| 100%
#>
#> [2023-11-21 07:44:15.497146] DEtest done
#> Elapsed time:7.23 secs
#> Use the DE features from AllMarkers_wilcox to calculate distance metric.
#> DE features number of the query data: 2000
#> [2023-11-21 07:44:15.795602] Start DEtest
#> Workers: 2
#> Find all markers(wilcox) among 13 groups...
#>
|
| | 0%
#>
#>
#>
#>
#>
#>
#>
#>
|
|=========================================================== | 54%
#>
#>
#>
#>
#>
#>
|
|==============================================================================================================| 100%
#>
#> [2023-11-21 07:44:40.172202] DEtest done
#> Elapsed time:24.38 secs
#> Use the DE features from AllMarkers_wilcox to calculate distance metric.
#> DE features number of the ref data: 650
#> Use 181 features to calculate distance.
#> Detected query data type: log_normalized_counts
#> Detected reference data type: log_normalized_counts
#> Calculate similarity...
#> Use 'raw' method to find neighbors.
#> Predict cell type...
#> Error: No cell overlap between new meta data and Seurat object
CellDimPlot(pancreas_sub, group.by = "KNNPredict_classification", label = TRUE)
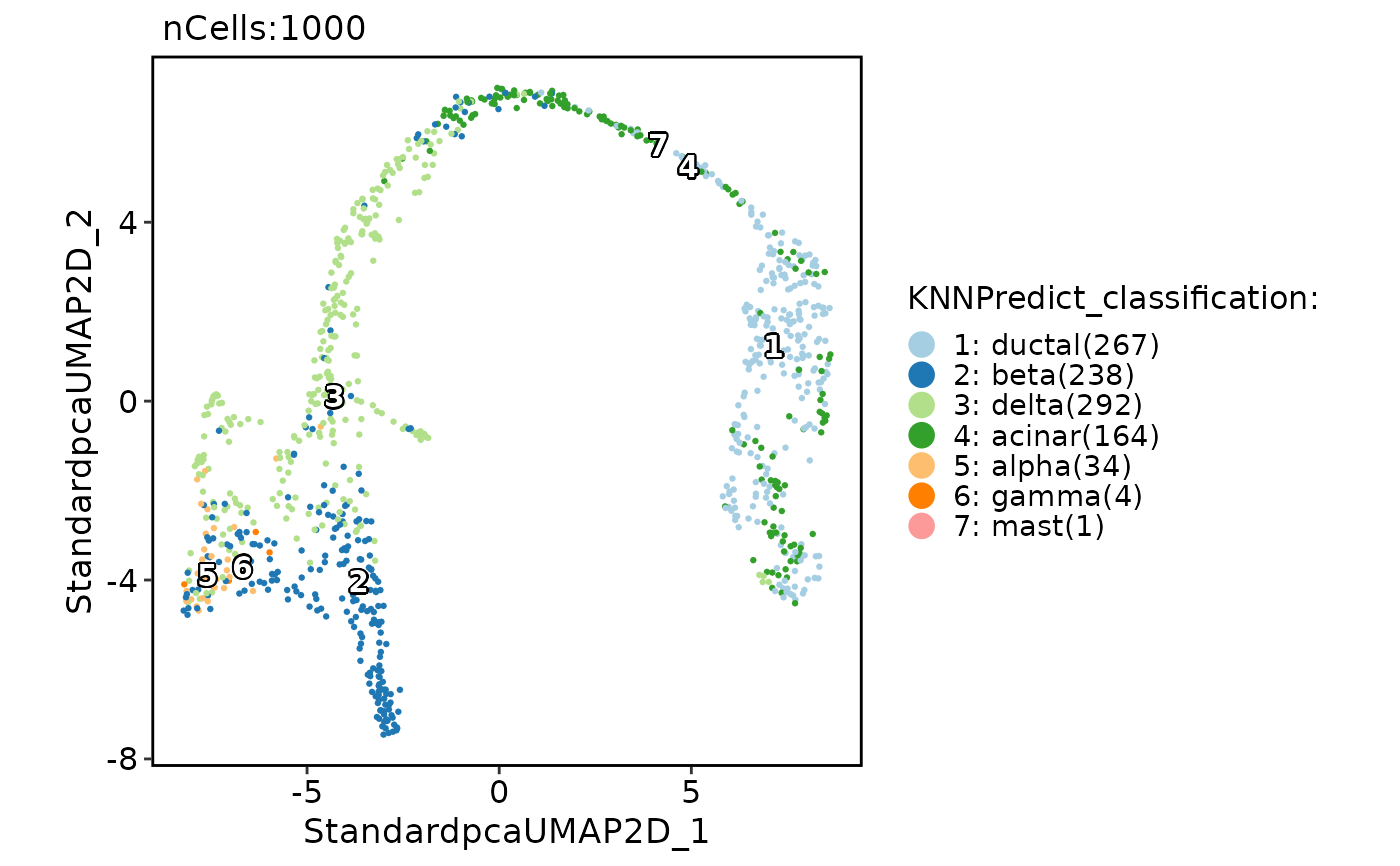 FeatureDimPlot(pancreas_sub, features = "KNNPredict_simil")
FeatureDimPlot(pancreas_sub, features = "KNNPredict_simil")